

Aortic Valve Embryology, Mechanobiology, and Second Messenger Pathways: Implications for Clinical Practice
 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
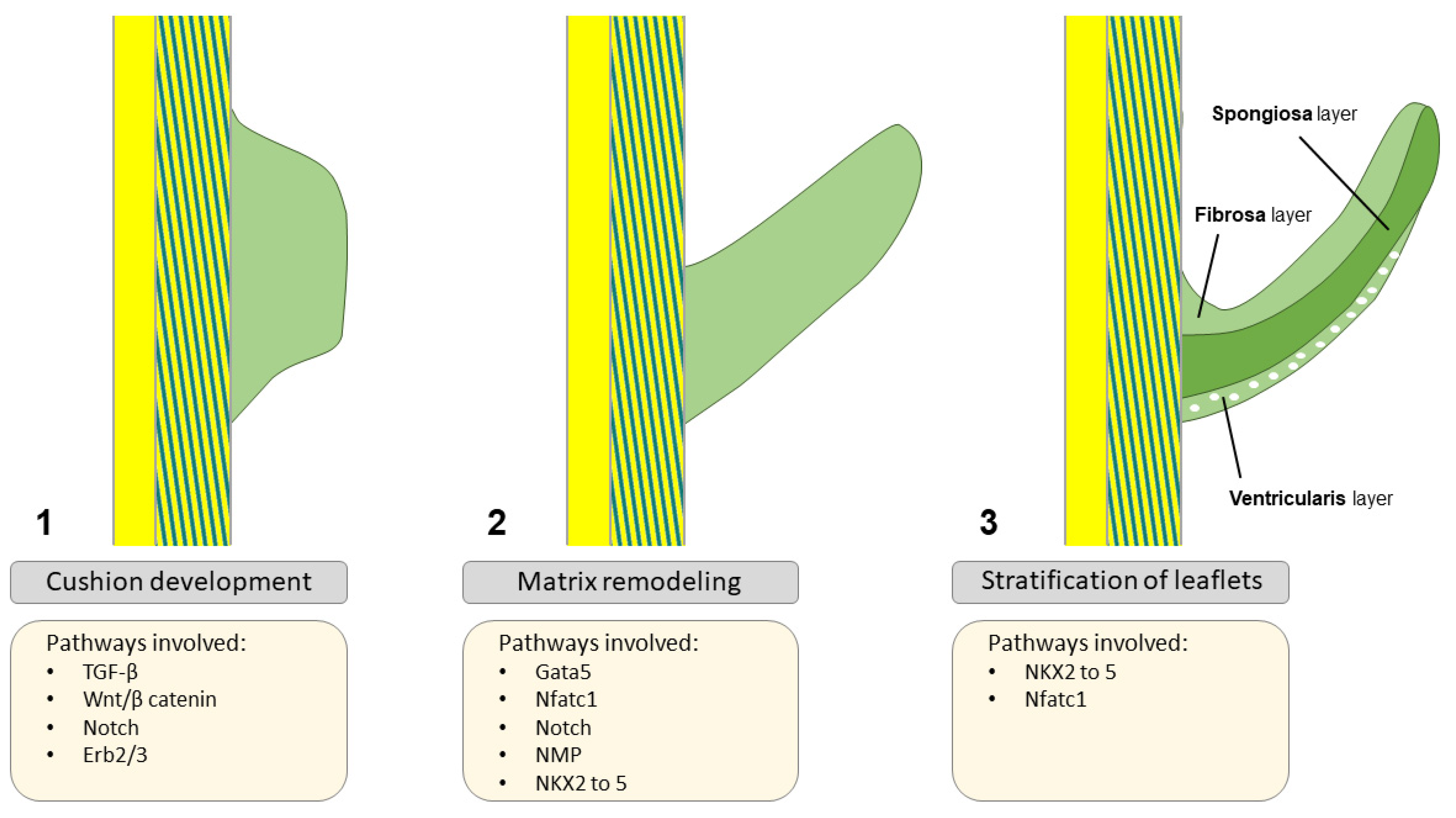
2. Embryology of the Aortic Valve
Normal Outflow Tract and Valve Formation
3. Transcriptional Regulation of Valvulogenesis
Timing/Expression of Transcription Factors and Deficient Aortic Valve Formation
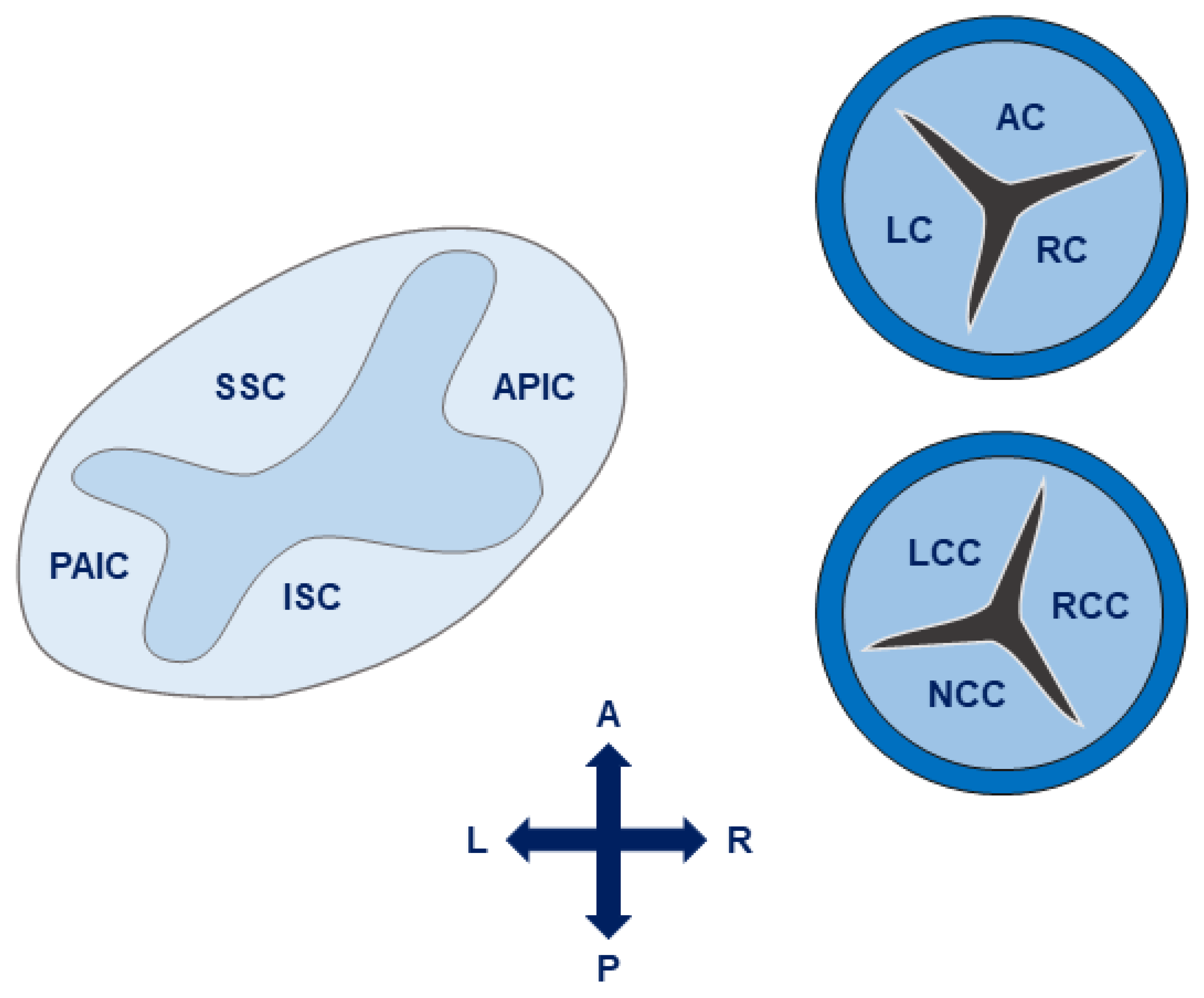
4. Anatomy and Hemodynamics
4.1. The Aortic Valve and Root
4.2. Valvular Fluid Dynamics
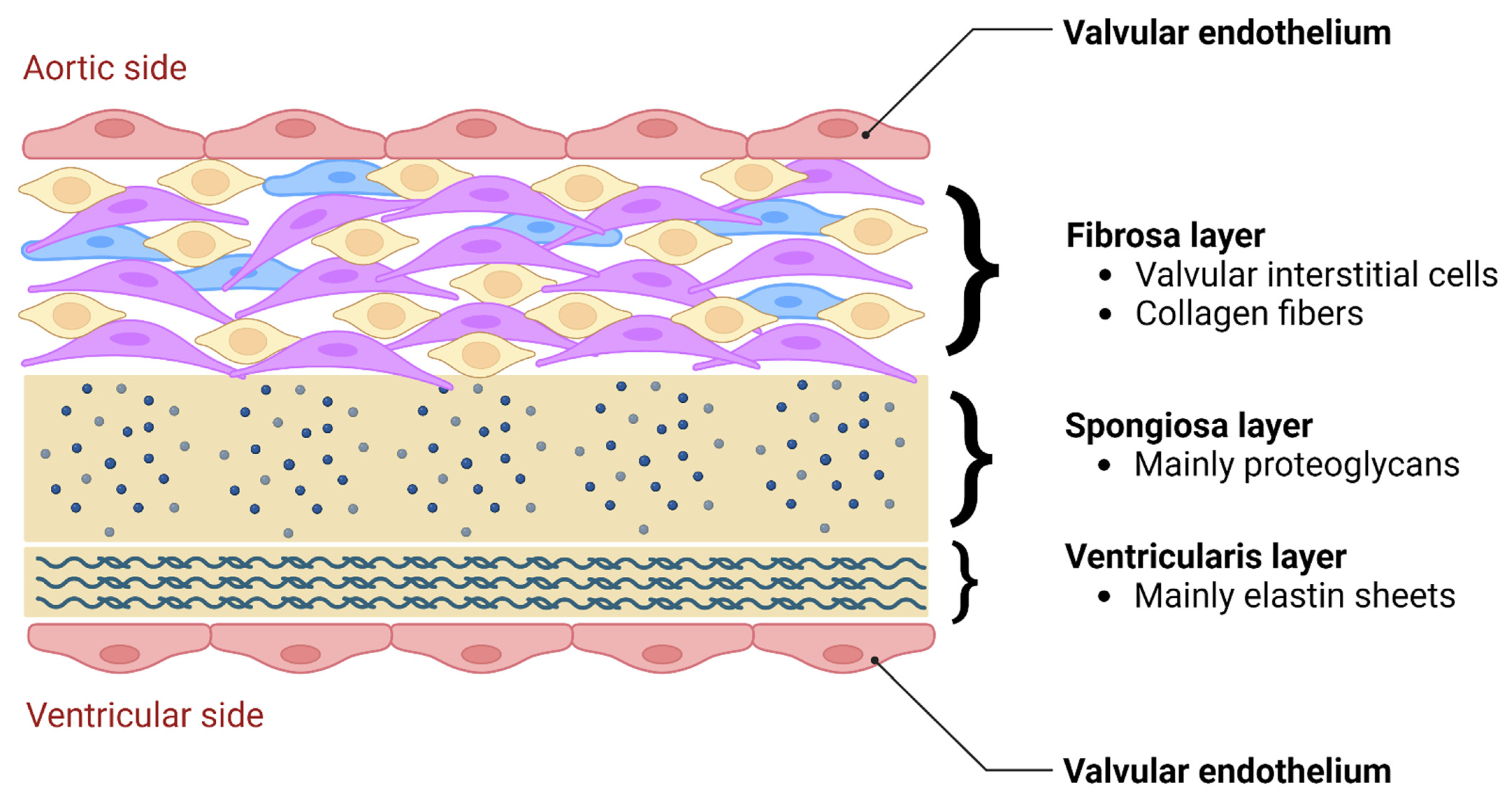
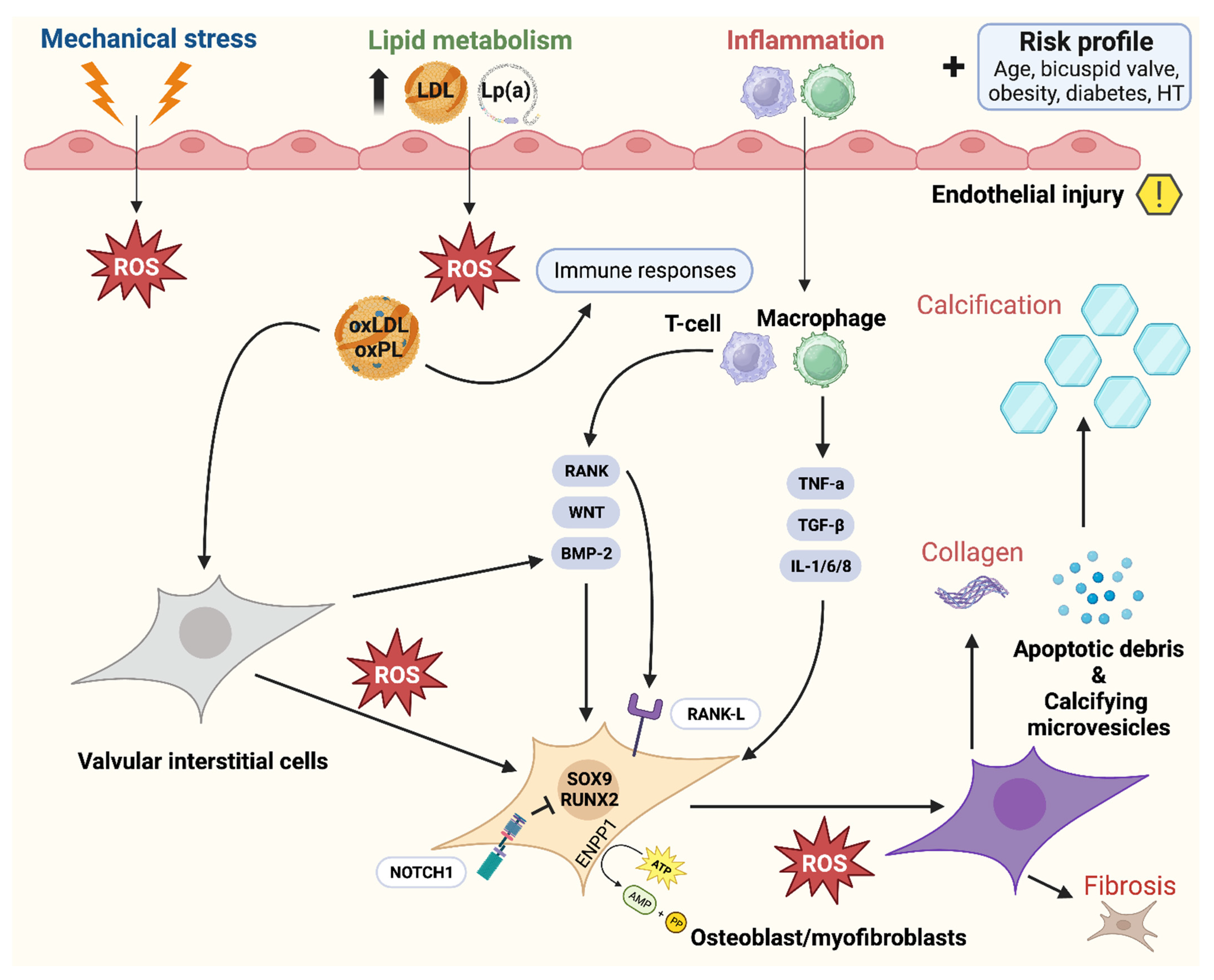
5. Biomechanics and Cellular Responses
5.1. Functional Morphology and Mechanical Stimuli
5.2. Calcific Aortic Valve Disease
5.3. Adaptive Remodeling
6. (Surgical) Treatment
6.1. Evidence-Based Medicine
6.2. Clinical Decision Making
7. Conclusions and Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- d’Arcy, J.L.; Prendergast, B.D.; Chambers, J.B.; Ray, S.G.; Bridgewater, B. Valvular heart disease: The next cardiac epidemic. Heart 2011, 97, 91–93. [Google Scholar] [CrossRef]
- Coffey, S.; Cox, B.; Williams, M.J. Lack of progress in valvular heart disease in the pre-transcatheter aortic valve replacement era: Increasing deaths and minimal change in mortality rate over the past three decades. Am. Heart J. 2014, 167, 562–567.e2. [Google Scholar] [CrossRef]
- Rosenhek, R.; Zilberszac, R.; Schemper, M.; Czerny, M.; Mundigler, G.; Graf, S.; Bergler-Klein, J.; Grimm, M.; Gabriel, H.; Maurer, G. Natural history of very severe aortic stenosis. Circulation 2010, 121, 151–156. [Google Scholar] [CrossRef] [PubMed]
- Ross, J., Jr.; Braunwald, E. Aortic stenosis. Circulation 1968, 38, 61–67. [Google Scholar] [CrossRef] [PubMed]
- Philippe, G.; Rahul, P.S.; Robert, J.C.; Lucy, A.; Omar, M.A.; Konstantinos, P.K.; Leo, M.; Mostafa, N.; Samir, R.K.; Rajendra, R.M.; et al. The Mortality Burden of Untreated Aortic Stenosis. J. Am. Coll. Cardiol. 2023, 82, 2101–2109. [Google Scholar] [CrossRef]
- Goldbarg, S.H.; Elmariah, S.; Miller, M.A.; Fuster, V. Insights into Degenerative Aortic Valve Disease. J. Am. Coll. Cardiol. 2007, 50, 1205–1213. [Google Scholar] [CrossRef] [PubMed]
- El-Hamamsy, I.; Chester, A.H.; Yacoub, M.H. Cellular regulation of the structure and function of aortic valves. J. Adv. Res. 2010, 1, 5–12. [Google Scholar] [CrossRef]
- El-Hamamsy, I.; Yacoub, M.H.; Chester, A.H. Neuronal regulation of aortic valve cusps. Curr. Vasc. Pharmacol. 2009, 7, 40–46. [Google Scholar] [CrossRef]
- El-Hamamsy, I.; Balachandran, K.; Yacoub, M.H.; Stevens, L.M.; Sarathchandra, P.; Taylor, P.M.; Yoganathan, A.P.; Chester, A.H. Endothelium-dependent regulation of the mechanical properties of aortic valve cusps. J. Am. Coll. Cardiol. 2009, 53, 1448–1455. [Google Scholar] [CrossRef]
- Boodhwani, M.; de Kerchove, L.; Glineur, D.; Poncelet, A.; Rubay, J.; Astarci, P.; Verhelst, R.; Noirhomme, P.; El Khoury, G. Repair-oriented classification of aortic insufficiency: Impact on surgical techniques and clinical outcomes. J. Thorac. Cardiovasc. Surg. 2009, 137, 286–294. [Google Scholar] [CrossRef]
- Misfeld, M.; Chester, A.H.; Sievers, H.H.; Yacoub, M.H. Biological mechanisms influencing the function of the aortic root. J. Card. Surg. 2002, 17, 363–368. [Google Scholar] [CrossRef]
- Chester, A.H.; El-Hamamsy, I.; Butcher, J.T.; Latif, N.; Bertazzo, S.; Yacoub, M.H. The living aortic valve: From molecules to function. Glob. Cardiol. Sci. Pract. 2014, 2014, 52–77. [Google Scholar] [CrossRef]
- Ross, D.N. Replacement of aortic and mitral valves with a pulmonary autograft. Lancet 1967, 2, 956–958. [Google Scholar] [CrossRef]
- Lansac, E.; de Kerchove, L. Aortic valve repair techniques: State of the art. Eur. J. Cardiothorac. Surg. 2018, 53, 1101–1107. [Google Scholar] [CrossRef]
- Combs, M.D.; Yutzey, K.E. Heart valve development: Regulatory networks in development and disease. Circ. Res. 2009, 105, 408–421. [Google Scholar] [CrossRef] [PubMed]
- Gittenberger-de Groot, A.C.; Bartelings, M.M.; Deruiter, M.C.; Poelmann, R.E. Basics of cardiac development for the understanding of congenital heart malformations. Pediatr. Res. 2005, 57, 169–176. [Google Scholar] [CrossRef] [PubMed]
- Moorman, A.F.; Christoffels, V.M. Cardiac chamber formation: Development, genes, and evolution. Physiol. Rev. 2003, 83, 1223–1267. [Google Scholar] [CrossRef] [PubMed]
- Lamers, W.H.; Moorman, A.F. Cardiac septation: A late contribution of the embryonic primary myocardium to heart morphogenesis. Circ. Res. 2002, 91, 93–103. [Google Scholar] [CrossRef]
- Soufan, A.T.; van den Hoff, M.J.; Ruijter, J.M.; de Boer, P.A.; Hagoort, J.; Webb, S.; Anderson, R.H.; Moorman, A.F. Reconstruction of the patterns of gene expression in the developing mouse heart reveals an architectural arrangement that facilitates the understanding of atrial malformations and arrhythmias. Circ. Res. 2004, 95, 1207–1215. [Google Scholar] [CrossRef] [PubMed]
- Schroeder, J.A.; Jackson, L.F.; Lee, D.C.; Camenisch, T.D. Form and function of developing heart valves: Coordination by extracellular matrix and growth factor signaling. J. Mol. Med. 2003, 81, 392–403. [Google Scholar] [CrossRef] [PubMed]
- Wirrig, E.E.; Yutzey, K.E. Conserved transcriptional regulatory mechanisms in aortic valve development and disease. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 737–741. [Google Scholar] [CrossRef]
- Hinton, R.B., Jr.; Lincoln, J.; Deutsch, G.H.; Osinska, H.; Manning, P.B.; Benson, D.W.; Yutzey, K.E. Extracellular matrix remodeling and organization in developing and diseased aortic valves. Circ. Res. 2006, 98, 1431–1438. [Google Scholar] [CrossRef]
- Martin, P.S.; Kloesel, B.; Norris, R.A.; Lindsay, M.; Milan, D.; Body, S.C. Embryonic Development of the Bicuspid Aortic Valve. J. Cardiovasc. Dev. Dis. 2015, 2, 248–272. [Google Scholar] [CrossRef]
- Rabkin-Aikawa, E.; Farber, M.; Aikawa, M.; Schoen, F.J. Dynamic and reversible changes of interstitial cell phenotype during remodeling of cardiac valves. J. Heart Valve Dis. 2004, 13, 841–847. [Google Scholar]
- de Lange, F.J.; Moorman, A.F.; Anderson, R.H.; Männer, J.; Soufan, A.T.; de Gier-de Vries, C.; Schneider, M.D.; Webb, S.; van den Hoff, M.J.; Christoffels, V.M. Lineage and morphogenetic analysis of the cardiac valves. Circ. Res. 2004, 95, 645–654. [Google Scholar] [CrossRef]
- Butcher, J.T.; Markwald, R.R. Valvulogenesis: The moving target. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2007, 362, 1489–1503. [Google Scholar] [CrossRef] [PubMed]
- Gobergs, R.; Salputra, E.; Lubaua, I. Hypoplastic left heart syndrome: A review. Acta Med. Litu. 2016, 23, 86–98. [Google Scholar] [CrossRef] [PubMed]
- Rahman, A.; DeYoung, T.; Cahill, L.S.; Yee, Y.; Debebe, S.K.; Botelho, O.; Seed, M.; Chaturvedi, R.R.; Sled, J.G. A mouse model of hypoplastic left heart syndrome demonstrating left heart hypoplasia and retrograde aortic arch flow. Dis. Models Mech. 2021, 14, dmm049077. [Google Scholar] [CrossRef] [PubMed]
- Greenberg, H.Z.E.; Zhao, G.; Shah, A.M.; Zhang, M. Role of oxidative stress in calcific aortic valve disease and its therapeutic implications. Cardiovasc. Res. 2021, 118, 1433–1451. [Google Scholar] [CrossRef]
- Dayawansa, N.H.; Baratchi, S.; Peter, K. Uncoupling the Vicious Cycle of Mechanical Stress and Inflammation in Calcific Aortic Valve Disease. Front. Cardiovasc. Med. 2022, 9, 783543. [Google Scholar] [CrossRef] [PubMed]
- Peng, Q.; Shan, D.; Cui, K.; Li, K.; Zhu, B.; Wu, H.; Wang, B.; Wong, S.; Norton, V.; Dong, Y.; et al. The Role of Endothelial-to-Mesenchymal Transition in Cardiovascular Disease. Cells 2022, 11, 1834. [Google Scholar] [CrossRef]
- Henderson, D.J.; Eley, L.; Turner, J.E.; Chaudhry, B. Development of the Human Arterial Valves: Understanding Bicuspid Aortic Valve. Front. Cardiovasc. Med. 2021, 8, 802930. [Google Scholar] [CrossRef] [PubMed]
- Hutson, M.R.; Kirby, M.L. Model systems for the study of heart development and disease: Cardiac neural crest and conotruncal malformations. Semin. Cell Dev. Biol. 2007, 18, 101–110. [Google Scholar] [CrossRef]
- George, R.M.; Maldonado-Velez, G.; Firulli, A.B. The heart of the neural crest: Cardiac neural crest cells in development and regeneration. Development 2020, 147, dev188706. [Google Scholar] [CrossRef] [PubMed]
- Poelmann, R.E.; Mikawa, T.; Gittenberger-de Groot, A.C. Neural crest cells in outflow tract septation of the embryonic chicken heart: Differentiation and apoptosis. Dev. Dyn. 1998, 212, 373–384. [Google Scholar] [CrossRef]
- Chakraborty, S.; Combs, M.D.; Yutzey, K.E. Transcriptional regulation of heart valve progenitor cells. Pediatr. Cardiol. 2010, 31, 414–421. [Google Scholar] [CrossRef] [PubMed]
- Maleki, S.; Poujade, F.A.; Bergman, O.; Gådin, J.R.; Simon, N.; Lång, K.; Franco-Cereceda, A.; Body, S.C.; Björck, H.M.; Eriksson, P. Endothelial/Epithelial Mesenchymal Transition in Ascending Aortas of Patients With Bicuspid Aortic Valve. Front. Cardiovasc. Med. 2019, 6, 182. [Google Scholar] [CrossRef]
- Armstrong, E.J.; Bischoff, J. Heart valve development: Endothelial cell signaling and differentiation. Circ. Res. 2004, 95, 459–470. [Google Scholar] [CrossRef]
- Camenisch, T.D.; Molin, D.G.; Person, A.; Runyan, R.B.; Gittenberger-de Groot, A.C.; McDonald, J.A.; Klewer, S.E. Temporal and distinct TGFbeta ligand requirements during mouse and avian endocardial cushion morphogenesis. Dev. Biol. 2002, 248, 170–181. [Google Scholar] [CrossRef]
- Ayoub, S.; Ferrari, G.; Gorman, R.C.; Gorman, J.H.; Schoen, F.J.; Sacks, M.S. Heart Valve Biomechanics and Underlying Mechanobiology. Compr. Physiol. 2016, 6, 1743–1780. [Google Scholar]
- Rochais, F.; Mesbah, K.; Kelly, R.G. Signaling pathways controlling second heart field development. Circ. Res. 2009, 104, 933–942. [Google Scholar] [CrossRef]
- Grewal, N.; Gittenberger-de Groot, A.C.; Lindeman, J.H.; Klautz, A.; Driessen, A.; Klautz, R.J.M.; Poelmann, R.E. Normal and abnormal development of the aortic valve and ascending aortic wall: A comprehensive overview of the embryology and pathology of the bicuspid aortic valve. Ann. Cardiothorac. Surg. 2022, 11, 380–388. [Google Scholar] [CrossRef] [PubMed]
- Harmon, A.W.; Nakano, A. Nkx2-5 lineage tracing visualizes the distribution of second heart field-derived aortic smooth muscle. Genesis 2013, 51, 862–869. [Google Scholar] [CrossRef] [PubMed]
- Sawada, H.; Rateri, D.L.; Moorleghen, J.J.; Majesky, M.W.; Daugherty, A. Smooth Muscle Cells Derived from Second Heart Field and Cardiac Neural Crest Reside in Spatially Distinct Domains in the Media of the Ascending Aorta-Brief Report. Arter. Thromb. Vasc. Biol. 2017, 37, 1722–1726. [Google Scholar] [CrossRef] [PubMed]
- George, V.; Colombo, S.; Targoff, K.L. An early requirement for nkx2.5 ensures the first and second heart field ventricular identity and cardiac function into adulthood. Dev. Biol. 2015, 400, 10–22. [Google Scholar] [CrossRef] [PubMed]
- Garside, V.C.; Chang, A.C.; Karsan, A.; Hoodless, P.A. Co-ordinating Notch, BMP, and TGF-β signaling during heart valve development. Cell. Mol. Life Sci. 2013, 70, 2899–2917. [Google Scholar] [CrossRef] [PubMed]
- Walker, G.A.; Masters, K.S.; Shah, D.N.; Anseth, K.S.; Leinwand, L.A. Valvular myofibroblast activation by transforming growth factor-beta: Implications for pathological extracellular matrix remodeling in heart valve disease. Circ. Res. 2004, 95, 253–260. [Google Scholar] [CrossRef] [PubMed]
- Niessen, K.; Fu, Y.; Chang, L.; Hoodless, P.A.; McFadden, D.; Karsan, A. Slug is a direct Notch target required for initiation of cardiac cushion cellularization. J. Cell Biol. 2008, 182, 315–325. [Google Scholar] [CrossRef]
- Loomes, K.M.; Taichman, D.B.; Glover, C.L.; Williams, P.T.; Markowitz, J.E.; Piccoli, D.A.; Baldwin, H.S.; Oakey, R.J. Characterization of Notch receptor expression in the developing mammalian heart and liver. Am. J. Med. Genet. 2002, 112, 181–189. [Google Scholar] [CrossRef]
- Varadkar, P.; Kraman, M.; Despres, D.; Ma, G.; Lozier, J.; McCright, B. Notch2 is required for the proliferation of cardiac neural crest-derived smooth muscle cells. Dev. Dyn. 2008, 237, 1144–1152. [Google Scholar] [CrossRef]
- Timmerman, L.A.; Grego-Bessa, J.; Raya, A.; Bertrán, E.; Pérez-Pomares, J.M.; Díez, J.; Aranda, S.; Palomo, S.; McCormick, F.; Izpisúa-Belmonte, J.C.; et al. Notch promotes epithelial-mesenchymal transition during cardiac development and oncogenic transformation. Genes. Dev. 2004, 18, 99–115. [Google Scholar] [CrossRef] [PubMed]
- Garg, V.; Muth, A.N.; Ransom, J.F.; Schluterman, M.K.; Barnes, R.; King, I.N.; Grossfeld, P.D.; Srivastava, D. Mutations in NOTCH1 cause aortic valve disease. Nature 2005, 437, 270–274. [Google Scholar] [CrossRef] [PubMed]
- Kamath, B.M.; Bauer, R.C.; Loomes, K.M.; Chao, G.; Gerfen, J.; Hutchinson, A.; Hardikar, W.; Hirschfield, G.; Jara, P.; Krantz, I.D.; et al. NOTCH2 mutations in Alagille syndrome. J. Med. Genet. 2012, 49, 138–144. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Chang, A.; Chang, L.; Niessen, K.; Eapen, S.; Setiadi, A.; Karsan, A. Differential regulation of transforming growth factor beta signaling pathways by Notch in human endothelial cells. J. Biol. Chem. 2009, 284, 19452–19462. [Google Scholar] [CrossRef] [PubMed]
- Kokubo, H.; Miyagawa-Tomita, S.; Tomimatsu, H.; Nakashima, Y.; Nakazawa, M.; Saga, Y.; Johnson, R.L. Targeted disruption of hesr2 results in atrioventricular valve anomalies that lead to heart dysfunction. Circ. Res. 2004, 95, 540–547. [Google Scholar] [CrossRef]
- Cripe, L.; Andelfinger, G.; Martin, L.J.; Shooner, K.; Benson, D.W. Bicuspid aortic valve is heritable. J. Am. Coll. Cardiol. 2004, 44, 138–143. [Google Scholar] [CrossRef] [PubMed]
- Tessler, I.; Albuisson, J.; Piñeiro-Sabarís, R.; Verstraeten, A.; Kamber Kaya, H.E.; Siguero-Álvarez, M.; Goudot, G.; MacGrogan, D.; Luyckx, I.; Shpitzen, S.; et al. Novel Association of the NOTCH Pathway Regulator MIB1 Gene with the Development of Bicuspid Aortic Valve. JAMA Cardiol. 2023, 8, 721–731. [Google Scholar] [CrossRef] [PubMed]
- MacGrogan, D.; D’Amato, G.; Travisano, S.; Martinez-Poveda, B.; Luxán, G.; Del Monte-Nieto, G.; Papoutsi, T.; Sbroggio, M.; Bou, V.; Gomez-Del Arco, P.; et al. Sequential Ligand-Dependent Notch Signaling Activation Regulates Valve Primordium Formation and Morphogenesis. Circ. Res. 2016, 118, 1480–1497. [Google Scholar] [CrossRef]
- Schlotter, F.; Halu, A.; Goto, S.; Blaser, M.C.; Body, S.C.; Lee, L.H.; Higashi, H.; DeLaughter, D.M.; Hutcheson, J.D.; Vyas, P.; et al. Spatiotemporal Multi-Omics Mapping Generates a Molecular Atlas of the Aortic Valve and Reveals Networks Driving Disease. Circulation 2018, 138, 377–393. [Google Scholar] [CrossRef]
- Ljungberg, J.; Janiec, M.; Bergdahl, I.A.; Holmgren, A.; Hultdin, J.; Johansson, B.; Näslund, U.; Siegbahn, A.; Fall, T.; Söderberg, S. Proteomic Biomarkers for Incident Aortic Stenosis Requiring Valvular Replacement. Circulation 2018, 138, 590–599. [Google Scholar] [CrossRef]
- Bourgeois, R.; Bourgault, J.; Despres, A.-A.; Perrot, N.; Guertin, J.; Girard, A.; Mitchell, P.L.; Gotti, C.; Bourassa, S.; Scipione, C.A.; et al. Lipoprotein Proteomics and Aortic Valve Transcriptomics Identify Biological Pathways Linking Lipoprotein(a) Levels to Aortic Stenosis. Metabolites 2021, 11, 459. [Google Scholar] [CrossRef] [PubMed]
- Fu, B.; Wang, J.; Wang, L.; Wang, Q.; Guo, Z.; Xu, M.; Jiang, N. Integrated proteomic and metabolomic profile analyses of cardiac valves revealed molecular mechanisms and targets in calcific aortic valve disease. Front. Cardiovasc. Med. 2022, 9, 944521. [Google Scholar] [CrossRef]
- Cahalane, R.A.; Clift, C.L.; Turner, M.E.; Blaser, M.C.; Kasai, T.; Hendrickx, A.; Campedelli, A.; Billaud, M.; Rega, F.; McNamara, L.; et al. Distinct Regulatory Mechanisms of Bioprosthetic And Native Aortic Valve Calcification: A Proteomic Comparison. In Proceedings of the Heart Valve Society Annual Meeting 2024, Boston, MA, USA, 19 February 2024. [Google Scholar]
- Small, A.M.; Peloso, G.M.; Linefsky, J.; Aragam, J.; Galloway, A.; Tanukonda, V.; Wang, L.C.; Yu, Z.; Sunitha Selvaraj, M.; Farber-Eger, E.H.; et al. Multiancestry Genome-Wide Association Study of Aortic Stenosis Identifies Multiple Novel Loci in the Million Veteran Program. Circulation 2023, 147, 942–955. [Google Scholar] [CrossRef]
- Gehlen, J.; Stundl, A.; Debiec, R.; Fontana, F.; Krane, M.; Sharipova, D.; Nelson, C.P.; Al-Kassou, B.; Giel, A.-S.; Sinning, J.-M.; et al. Elucidation of the genetic causes of bicuspid aortic valve disease. Cardiovasc. Res. 2022, 119, 857–866. [Google Scholar] [CrossRef] [PubMed]
- Yap, C.H.; Saikrishnan, N.; Yoganathan, A.P. Experimental measurement of dynamic fluid shear stress on the ventricular surface of the aortic valve leaflet. Biomech. Model. Mechanobiol. 2012, 11, 231–244. [Google Scholar] [CrossRef]
- Yacoub, M.H.; Kilner, P.J.; Birks, E.J.; Misfeld, M. The aortic outflow and root: A tale of dynamism and crosstalk. Ann. Thorac. Surg. 1999, 68, S37–S43. [Google Scholar] [CrossRef] [PubMed]
- Anderson, R.H. Clinical anatomy of the aortic root. Heart 2000, 84, 670–673. [Google Scholar] [CrossRef]
- Anderson, R.H. The surgical anatomy of the aortic root. Multimed. Man. Cardiothorac. Surg. MMCTS 2007, 2007, mmcts.2006.002527. [Google Scholar] [CrossRef]
- Sarsam, M.A.; Yacoub, M. Remodeling of the aortic valve anulus. J. Thorac. Cardiovasc. Surg. 1993, 105, 435–438. [Google Scholar] [CrossRef]
- David, T.E.; Feindel, C.M. An aortic valve-sparing operation for patients with aortic incompetence and aneurysm of the ascending aorta. J. Thorac. Cardiovasc. Surg. 1992, 103, 617–621; discussion 622. [Google Scholar] [CrossRef]
- Rajamannan, N.M. Bicuspid aortic valve disease: The role of oxidative stress in Lrp5 bone formation. Cardiovasc. Pathol. 2011, 20, 168–176. [Google Scholar] [CrossRef]
- Fries, R.; Graeter, T.; Aicher, D.; Reul, H.; Schmitz, C.; Böhm, M.; Schäfers, H.-J. In Vitro comparison of aortic valve movement after valve-preserving aortic replacement. J. Thorac. Cardiovasc. Surg. 2006, 132, 32–37. [Google Scholar] [CrossRef]
- Arjunon, S.; Rathan, S.; Jo, H.; Yoganathan, A.P. Aortic valve: Mechanical environment and mechanobiology. Ann. Biomed. Eng. 2013, 41, 1331–1346. [Google Scholar] [CrossRef] [PubMed]
- Bellhouse, B.J.; Talbot, L. The fluid mechanics of the aortic valve. J. Fluid Mech. 1969, 35, 721–735. [Google Scholar] [CrossRef]
- Campinho, P.; Vilfan, A.; Vermot, J. Blood Flow Forces in Shaping the Vascular System: A Focus on Endothelial Cell Behavior. Front. Physiol. 2020, 11, 552. [Google Scholar] [CrossRef] [PubMed]
- Yacoub, M.H.; Aguib, H.; Gamrah, M.A.; Shehata, N.; Nagy, M.; Donia, M.; Aguib, Y.; Saad, H.; Romeih, S.; Torii, R.; et al. Aortic root dynamism, geometry, and function after the remodeling operation: Clinical relevance. J. Thorac. Cardiovasc. Surg. 2018, 156, 951–962.e2. [Google Scholar] [CrossRef] [PubMed]
- Bellhouse, B.; Bellhouse, F. Fluid mechanics of model normal and stenosed aortic valves. Circ. Res. 1969, 25, 693–704. [Google Scholar] [PubMed]
- Hamdan, A.; Guetta, V.; Konen, E.; Goitein, O.; Segev, A.; Raanani, E.; Spiegelstein, D.; Hay, I.; Di Segni, E.; Eldar, M.; et al. Deformation dynamics and mechanical properties of the aortic annulus by 4-dimensional computed tomography: Insights into the functional anatomy of the aortic valve complex and implications for transcatheter aortic valve therapy. J. Am. Coll. Cardiol. 2012, 59, 119–127. [Google Scholar] [CrossRef] [PubMed]
- Cheng, A.; Dagum, P.; Miller, D.C. Aortic root dynamics and surgery: From craft to science. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2007, 362, 1407–1419. [Google Scholar] [CrossRef] [PubMed]
- Bruno, R.M.; Climie, R.; Gallo, A. Aortic pulsatility drives microvascular organ damage in essential hypertension: New evidence from choroidal thickness assessment. J. Clin. Hypertens. 2021, 23, 1039–1040. [Google Scholar] [CrossRef]
- Um, K.J.; McClure, G.R.; Belley-Cote, E.P.; Gupta, S.; Bouhout, I.; Lortie, H.; Alraddadi, H.; Alsagheir, A.; Bossard, M.; McIntyre, W.F.; et al. Hemodynamic outcomes of the Ross procedure versus other aortic valve replacement: A systematic review and meta-analysis. J. Cardiovasc. Surg. 2018, 59, 462–470. [Google Scholar] [CrossRef]
- Stewart, S.; Afoakwah, C.; Chan, Y.-K.; Strom, J.B.; Playford, D.; Strange, G.A. Counting the cost of premature mortality with progressively worse aortic stenosis in Australia: A clinical cohort study. Lancet Healthy Longev. 2022, 3, e599–e606. [Google Scholar] [CrossRef]
- Harris, P.; Kuppurao, L. Quantitative Doppler echocardiography. BJA Educ. 2015, 16, 46–52. [Google Scholar] [CrossRef]
- Chan, V.; Rubens, F.; Boodhwani, M.; Mesana, T.; Ruel, M. Determinants of persistent or recurrent congestive heart failure after contemporary surgical aortic valve replacement. J. Heart Valve Dis. 2014, 23, 665–670. [Google Scholar] [PubMed]
- Duebener, L.F.; Stierle, U.; Erasmi, A.; Bechtel, M.F.; Zurakowski, D.; Böhm, J.O.; Botha, C.A.; Hemmer, W.; Rein, J.G.; Sievers, H.H.; et al. Ross procedure and left ventricular mass regression. Circulation 2005, 112, I415–I422. [Google Scholar] [CrossRef] [PubMed]
- Hauser, M.; Bengel, F.M.; Kühn, A.; Sauer, U.; Zylla, S.; Braun, S.L.; Nekolla, S.G.; Oberhoffer, R.; Lange, R.; Schwaiger, M.; et al. Myocardial blood flow and flow reserve after coronary reimplantation in patients after arterial switch and ross operation. Circulation 2001, 103, 1875–1880. [Google Scholar] [CrossRef] [PubMed]
- Gaudino, M.; Piatti, F.; Lau, C.; Sturla, F.; Weinsaft, J.W.; Weltert, L.; Votta, E.; Galea, N.; Chirichilli, I.; Di Franco, A.; et al. Aortic flow after valve sparing root replacement with or without neosinuses reconstruction. J. Thorac. Cardiovasc. Surg. 2019, 157, 455–465. [Google Scholar] [CrossRef] [PubMed]
- Balachandran, K.; Sucosky, P.; Yoganathan, A.P. Hemodynamics and mechanobiology of aortic valve inflammation and calcification. Int. J. Inflamm. 2011, 2011, 263870. [Google Scholar] [CrossRef] [PubMed]
- Rego, B.V.; Sacks, M.S. A functionally graded material model for the transmural stress distribution of the aortic valve leaflet. J. Biomech. 2017, 54, 88–95. [Google Scholar] [CrossRef] [PubMed]
- Kodigepalli, K.M.; Thatcher, K.; West, T.; Howsmon, D.P.; Schoen, F.J.; Sacks, M.S.; Breuer, C.K.; Lincoln, J. Biology and Biomechanics of the Heart Valve Extracellular Matrix. J. Cardiovasc. Dev. Dis. 2020, 7, 57. [Google Scholar] [CrossRef]
- Butcher, J.T.; Penrod, A.M.; García, A.J.; Nerem, R.M. Unique morphology and focal adhesion development of valvular endothelial cells in static and fluid flow environments. Arter. Thromb. Vasc. Biol. 2004, 24, 1429–1434. [Google Scholar] [CrossRef] [PubMed]
- Bäck, M.; Gasser, T.C.; Michel, J.-B.; Caligiuri, G. Biomechanical factors in the biology of aortic wall and aortic valve diseases. Cardiovasc. Res. 2013, 99, 232–241. [Google Scholar] [CrossRef] [PubMed]
- Farivar, R.S.; Cohn, L.H.; Soltesz, E.G.; Mihaljevic, T.; Rawn, J.D.; Byrne, J.G. Transcriptional profiling and growth kinetics of endothelium reveals differences between cells derived from porcine aorta versus aortic valve. Eur. J. Cardiothorac. Surg. 2003, 24, 527–534. [Google Scholar] [CrossRef] [PubMed]
- Simmons, C.A.; Grant, G.R.; Manduchi, E.; Davies, P.F. Spatial heterogeneity of endothelial phenotypes correlates with side-specific vulnerability to calcification in normal porcine aortic valves. Circ. Res. 2005, 96, 792–799. [Google Scholar] [CrossRef]
- Butcher, J.T.; Tressel, S.; Johnson, T.; Turner, D.; Sorescu, G.; Jo, H.; Nerem, R.M. Transcriptional profiles of valvular and vascular endothelial cells reveal phenotypic differences: Influence of shear stress. Arter. Thromb. Vasc. Biol. 2006, 26, 69–77. [Google Scholar] [CrossRef]
- Yu Chen, H.; Dina, C.; Small, A.M.; Shaffer, C.M.; Levinson, R.T.; Helgadóttir, A.; Capoulade, R.; Munter, H.M.; Martinsson, A.; Cairns, B.J.; et al. Dyslipidemia, inflammation, calcification, and adiposity in aortic stenosis: A genome-wide study. Eur. Heart J. 2023, 44, 1927–1939. [Google Scholar] [CrossRef]
- Scott, A.J.; Simon, L.R.; Hutson, H.N.; Porras, A.M.; Masters, K.S. Engineering the aortic valve extracellular matrix through stages of development, aging, and disease. J. Mol. Cell. Cardiol. 2021, 161, 1–8. [Google Scholar] [CrossRef]
- El-Hamamsy, I.; Yacoub, M.H. Cellular and molecular mechanisms of thoracic aortic aneurysms. Nat. Rev. Cardiol. 2009, 6, 771–786. [Google Scholar] [CrossRef]
- Leopold, J.A. Cellular mechanisms of aortic valve calcification. Circ. Cardiovasc. Interv. 2012, 5, 605–614. [Google Scholar] [CrossRef]
- Chen, J.H.; Simmons, C.A. Cell-matrix interactions in the pathobiology of calcific aortic valve disease: Critical roles for matricellular, matricrine, and matrix mechanics cues. Circ. Res. 2011, 108, 1510–1524. [Google Scholar] [CrossRef]
- Yip, C.Y.; Chen, J.H.; Zhao, R.; Simmons, C.A. Calcification by valve interstitial cells is regulated by the stiffness of the extracellular matrix. Arter. Thromb. Vasc. Biol. 2009, 29, 936–942. [Google Scholar] [CrossRef]
- Lerman, D.A.; Prasad, S.; Alotti, N. Calcific Aortic Valve Disease: Molecular Mechanisms and Therapeutic Approaches. Eur. Cardiol. 2015, 10, 108–112. [Google Scholar] [CrossRef]
- Rajamannan, N.M.; Subramaniam, M.; Rickard, D.; Stock, S.R.; Donovan, J.; Springett, M.; Orszulak, T.; Fullerton, D.A.; Tajik, A.J.; Bonow, R.O.; et al. Human aortic valve calcification is associated with an osteoblast phenotype. Circulation 2003, 107, 2181–2184. [Google Scholar] [CrossRef]
- Jian, B.; Narula, N.; Li, Q.-y.; Mohler, E.R.; Levy, R.J. Progression of aortic valve stenosis: TGF-β1 is present in calcified aortic valve cusps and promotes aortic valve interstitial cell calcification via apoptosis. Ann. Thorac. Surg. 2003, 75, 457–465. [Google Scholar] [CrossRef]
- Goel, S.S.; Kleiman, N.S.; Zoghbi, W.A.; Reardon, M.J.; Kapadia, S.R. Renin-Angiotensin System Blockade in Aortic Stenosis: Implications Before and After Aortic Valve Replacement. J. Am. Heart Assoc. 2020, 9, e016911. [Google Scholar] [CrossRef]
- Thanassoulis, G.; Campbell, C.Y.; Owens, D.S.; Smith, J.G.; Smith, A.V.; Peloso, G.M.; Kerr, K.F.; Pechlivanis, S.; Budoff, M.J.; Harris, T.B.; et al. Genetic Associations with Valvular Calcification and Aortic Stenosis. N. Engl. J. Med. 2013, 368, 503–512. [Google Scholar] [CrossRef]
- Rosenhek, R.; Rader, F.; Loho, N.; Gabriel, H.; Heger, M.; Klaar, U.; Schemper, M.; Binder, T.; Maurer, G.; Baumgartner, H. Statins but not angiotensin-converting enzyme inhibitors delay progression of aortic stenosis. Circulation 2004, 110, 1291–1295. [Google Scholar] [CrossRef] [PubMed]
- Torzewski, M.; Ravandi, A.; Yeang, C.; Edel, A.; Bhindi, R.; Kath, S.; Twardowski, L.; Schmid, J.; Yang, X.; Franke, U.F.W.; et al. Lipoprotein(a) Associated Molecules are Prominent Components in Plasma and Valve Leaflets in Calcific Aortic Valve Stenosis. JACC Basic Transl. Sci. 2017, 2, 229–240. [Google Scholar] [CrossRef] [PubMed]
- Capoulade, R.; Chan, K.L.; Yeang, C.; Mathieu, P.; Bossé, Y.; Dumesnil, J.G.; Tam, J.W.; Teo, K.K.; Mahmut, A.; Yang, X.; et al. Oxidized Phospholipids, Lipoprotein(a), and Progression of Calcific Aortic Valve Stenosis. J. Am. Coll. Cardiol. 2015, 66, 1236–1246. [Google Scholar] [CrossRef] [PubMed]
- Cowell, S.J.; Newby, D.E.; Prescott, R.J.; Bloomfield, P.; Reid, J.; Northridge, D.B.; Boon, N.A. A Randomized Trial of Intensive Lipid-Lowering Therapy in Calcific Aortic Stenosis. N. Engl. J. Med. 2005, 352, 2389–2397. [Google Scholar] [CrossRef] [PubMed]
- Capoulade, R.; Clavel, M.A.; Dumesnil, J.G.; Chan, K.L.; Teo, K.K.; Tam, J.W.; Côté, N.; Mathieu, P.; Després, J.P.; Pibarot, P.; et al. Impact of metabolic syndrome on progression of aortic stenosis: Influence of age and statin therapy. J. Am. Coll. Cardiol. 2012, 60, 216–223. [Google Scholar] [CrossRef]
- Dichtl, W.; Alber, H.F.; Feuchtner, G.M.; Hintringer, F.; Reinthaler, M.; Bartel, T.; Süssenbacher, A.; Grander, W.; Ulmer, H.; Pachinger, O.; et al. Prognosis and risk factors in patients with asymptomatic aortic stenosis and their modulation by atorvastatin (20 mg). Am. J. Cardiol. 2008, 102, 743–748. [Google Scholar] [CrossRef] [PubMed]
- Gollmann-Tepeköylü, C.; Graber, M.; Hirsch, J.; Mair, S.; Naschberger, A.; Pölzl, L.; Nägele, F.; Kirchmair, E.; Degenhart, G.; Demetz, E.; et al. Toll-Like Receptor 3 Mediates Aortic Stenosis Through a Conserved Mechanism of Calcification. Circulation 2023, 147, 1518–1533. [Google Scholar] [CrossRef] [PubMed]
- Vahanian, A.; Beyersdorf, F.; Praz, F.; Milojevic, M.; Baldus, S.; Bauersachs, J.; Capodanno, D.; Conradi, L.; De Bonis, M.; De Paulis, R.; et al. 2021 ESC/EACTS Guidelines for the management of valvular heart disease: Developed by the Task Force for the management of valvular heart disease of the European Society of Cardiology (ESC) and the European Association for Cardio-Thoracic Surgery (EACTS). Eur. Heart J. 2021, 43, 561–632. [Google Scholar] [CrossRef] [PubMed]
- Moncla, L.M.; Briend, M.; Bossé, Y.; Mathieu, P. Calcific aortic valve disease: Mechanisms, prevention and treatment. Nat. Rev. Cardiol. 2023, 20, 546–559. [Google Scholar] [CrossRef] [PubMed]
- Khosla, S.; Eghbali-Fatourechi, G.Z. Circulating cells with osteogenic potential. Ann. N. Y. Acad. Sci. 2006, 1068, 489–497. [Google Scholar] [CrossRef] [PubMed]
- Egan, K.P.; Kim, J.H.; Mohler, E.R., 3rd; Pignolo, R.J. Role for circulating osteogenic precursor cells in aortic valvular disease. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 2965–2971. [Google Scholar] [CrossRef] [PubMed]
- Eghbali-Fatourechi, G.Z.; Lamsam, J.; Fraser, D.; Nagel, D.; Riggs, B.L.; Khosla, S. Circulating osteoblast-lineage cells in humans. N. Engl. J. Med. 2005, 352, 1959–1966. [Google Scholar] [CrossRef] [PubMed]
- Notenboom, M.L.; Schuermans, A.; Etnel, J.R.G.; Veen, K.M.; van de Woestijne, P.C.; Rega, F.R.; Helbing, W.A.; Bogers, A.; Takkenberg, J.J.M. Paediatric aortic valve replacement: A meta-analysis and microsimulation study. Eur. Heart J. 2023, 44, 3231–3246. [Google Scholar] [CrossRef]
- Schoof, P.H.; Cromme-Dijkhuis, A.H.; Bogers, J.J.; Thijssen, E.J.; Witsenburg, M.; Hess, J.; Bos, E. Aortic root replacement with pulmonary autograft in children. J. Thorac. Cardiovasc. Surg. 1994, 107, 367–373. [Google Scholar] [CrossRef]
- Van Hoof, L.; Verbrugghe, P.; Jones, E.A.V.; Humphrey, J.D.; Janssens, S.; Famaey, N.; Rega, F. Understanding Pulmonary Autograft Remodeling after the Ross Procedure: Stick to the Facts. Front. Cardiovasc. Med. 2022, 9, 829120. [Google Scholar] [CrossRef] [PubMed]
- Richardson, R.; Eley, L.; Donald-Wilson, C.; Davis, J.; Curley, N.; Alqahtani, A.; Murphy, L.; Anderson, R.H.; Henderson, D.J.; Chaudhry, B. Development and maturation of the fibrous components of the arterial roots in the mouse heart. J. Anat. 2018, 232, 554–567. [Google Scholar] [CrossRef] [PubMed]
- Jashari, R.; Van Hoeck, B.; Goffin, Y.; Vanderkelen, A. The incidence of congenital bicuspid or bileaflet and quadricuspid or quadrileaflet arterial valves in 3861 donor hearts in the European Homograft Bank. J. Heart Valve Dis. 2009, 18, 337–344. [Google Scholar]
- Rabkin-Aikawa, E.; Aikawa, M.; Farber, M.; Kratz, J.R.; Garcia-Cardena, G.; Kouchoukos, N.T.; Mitchell, M.B.; Jonas, R.A.; Schoen, F.J. Clinical pulmonary autograft valves: Pathologic evidence of adaptive remodeling in the aortic site. J. Thorac. Cardiovasc. Surg. 2004, 128, 552–561. [Google Scholar] [CrossRef]
- Mazine, A.; El-Hamamsy, I.; Verma, S.; Peterson, M.D.; Bonow, R.O.; Yacoub, M.H.; David, T.E.; Bhatt, D.L. Ross Procedure in Adults for Cardiologists and Cardiac Surgeons: JACC State-of-the-Art Review. J. Am. Coll. Cardiol. 2018, 72, 2761–2777. [Google Scholar] [CrossRef] [PubMed]
- Gorczynski, A.; Trenkner, M.; Anisimowicz, L.; Gutkowski, R.; Drapella, A.; Kwiatkowska, E.; Dobke, M. Biomechanics of the pulmonary autograft valve in the aortic position. Thorax 1982, 37, 535–539. [Google Scholar] [CrossRef]
- Xuan, Y.; Alonso, E.; Emmott, A.; Wang, Z.; Kumar, S.; Mongeon, F.-P.; Leask, R.L.; El-Hamamsy, I.; Ge, L.; Tseng, E.E. Wall stresses of early remodeled pulmonary autografts. J. Thorac. Cardiovasc. Surg. 2022, 164, 1728–1738.e2. [Google Scholar] [CrossRef]
- Mazine, A.; El-Hamamsy, I. The Ross procedure is an excellent operation in non-repairable aortic regurgitation: Insights and techniques. Ann. Cardiothorac. Surg. 2021, 10, 463–475. [Google Scholar] [CrossRef]
- Tanaka, D.; Mazine, A.; Ouzounian, M.; El-Hamamsy, I. Supporting the Ross procedure: Preserving root physiology while mitigating autograft dilatation. Curr. Opin. Cardiol. 2022, 37, 180–190. [Google Scholar] [CrossRef]
- Latif, N.; Mahgoub, A.; Nagy, M.; Sarathchandra, P.; Yacoub, M.H. Severe degeneration of a sub-coronary pulmonary autograft in a young adult. Glob. Cardiol. Sci. Pract. 2021, 2021, e202114. [Google Scholar] [CrossRef]
- Mokhles, S.; Takkenberg, J.J.M.; Treasure, T. Evidence-Based and Personalized Medicine. It’s [AND] not [OR]. Ann. Thorac. Surg. 2017, 103, 351–360. [Google Scholar] [CrossRef]
- David, L.S.; William, M.C.R.; Gray, J.A.M.; Haynes, R.B.; Richardson, W.S. Evidence based medicine: What it is and what it isn’t. BMJ 1996, 312, 71–72. [Google Scholar] [CrossRef]
- Etnel, J.R.G.; Huygens, S.A.; Grashuis, P.; Pekbay, B.; Papageorgiou, G.; Roos Hesselink, J.W.; Bogers, A.; Takkenberg, J.J.M. Bioprosthetic Aortic Valve Replacement in Nonelderly Adults: A Systematic Review, Meta-Analysis, Microsimulation. Circ. Cardiovasc. Qual. Outcomes 2019, 12, e005481. [Google Scholar] [CrossRef] [PubMed]
- Goldstone, A.B.; Chiu, P.; Baiocchi, M.; Lingala, B.; Patrick, W.L.; Fischbein, M.P.; Woo, Y.J. Mechanical or Biologic Prostheses for Aortic-Valve and Mitral-Valve Replacement. N. Engl. J. Med. 2017, 377, 1847–1857. [Google Scholar] [CrossRef]
- Korteland, N.M.; Etnel, J.R.G.; Arabkhani, B.; Mokhles, M.M.; Mohamad, A.; Roos-Hesselink, J.W.; Bogers, A.; Takkenberg, J.J.M. Mechanical aortic valve replacement in non-elderly adults: Meta-analysis and microsimulation. Eur. Heart J. 2017, 38, 3370–3377. [Google Scholar] [CrossRef]
- El-Hamamsy, I.; Eryigit, Z.; Stevens, L.M.; Sarang, Z.; George, R.; Clark, L.; Melina, G.; Takkenberg, J.J.; Yacoub, M.H. Long-term outcomes after autograft versus homograft aortic root replacement in adults with aortic valve disease: A randomised controlled trial. Lancet 2010, 376, 524–531. [Google Scholar] [CrossRef]
- Yacoub, M.; Rasmi, N.R.; Sundt, T.M.; Lund, O.; Boyland, E.; Radley-Smith, R.; Khaghani, A.; Mitchell, A. Fourteen-year experience with homovital homografts for aortic valve replacement. J. Thorac. Cardiovasc. Surg. 1995, 110, 186–193; discussion 184–193. [Google Scholar] [CrossRef] [PubMed]
- Notenboom, M.L.; Melina, G.; Veen, K.; De Robertis, F.; Coppola, G.; De Siena, P.; Navarra, E.; Gaer, J.; Ibrahim, M.; El-Hamamsy, I.; et al. Long-Term Clinical and Echocardiographic Outcomes Following the Ross Procedure: A Post Hoc Analysis of a Randomized Controlled Trial. JAMA Cardiol. 2023, in press. [CrossRef] [PubMed]
- Klieverik, L.M.; Takkenberg, J.J.; Bekkers, J.A.; Roos-Hesselink, J.W.; Witsenburg, M.; Bogers, A.J. The Ross operation: A Trojan horse? Eur. Heart J. 2007, 28, 1993–2000. [Google Scholar] [CrossRef]
- Reece, T.B.; Welke, K.F.; O’Brien, S.; Grau-Sepulveda, M.V.; Grover, F.L.; Gammie, J.S. Rethinking the ross procedure in adults. Ann. Thorac. Surg. 2014, 97, 175–181. [Google Scholar] [CrossRef]
- El-Hamamsy, I.; Toyoda, N.; Itagaki, S.; Stelzer, P.; Varghese, R.; Williams, E.E.; Erogova, N.; Adams, D.H. Propensity-Matched Comparison of the Ross Procedure and Prosthetic Aortic Valve Replacement in Adults. J. Am. Coll. Cardiol. 2022, 79, 805–815. [Google Scholar] [CrossRef]
- Ismail, E.-H.; Patrick, T.O.G.; David, H.A. The Ross Procedure. J. Am. Coll. Cardiol. 2022, 79, 1006–1009. [Google Scholar] [CrossRef]
- Yacoub, M.H.; Notenboom, M.L.; Melina, G.; Takkenberg, J.J.M. Surgical Heritage: You Had to be There, Ross: The Comeback Kid. In Seminars in Thoracic and Cardiovascular Surgery: Pediatric Cardiac Surgery Annual; Elsevier: Amsterdam, The Netherlands, 2023. [Google Scholar] [CrossRef]
- Tom, T.; Asif, H.; Magdi, Y. Is there a risk in avoiding risk for younger patients with aortic valve disease? BMJ 2011, 342, d2466. [Google Scholar] [CrossRef]
- Fioretta, E.S.; Motta, S.E.; Lintas, V.; Loerakker, S.; Parker, K.K.; Baaijens, F.P.T.; Falk, V.; Hoerstrup, S.P.; Emmert, M.Y. Next-generation tissue-engineered heart valves with repair, remodelling and regeneration capacity. Nat. Rev. Cardiol. 2021, 18, 92–116. [Google Scholar] [CrossRef]
- Huygens, S.A.; Rutten-van Mölken, M.; Noruzi, A.; Etnel, J.R.G.; Corro Ramos, I.; Bouten, C.V.C.; Kluin, J.; Takkenberg, J.J.M. What Is the Potential of Tissue-Engineered Pulmonary Valves in Children? Ann. Thorac. Surg. 2019, 107, 1845–1853. [Google Scholar] [CrossRef]
- van Haaften, E.E.; Bouten, C.V.C.; Kurniawan, N.A. Vascular Mechanobiology: Towards Control of In Situ Regeneration. Cells 2017, 6, 19. [Google Scholar] [CrossRef] [PubMed]
- Wissing, T.B.; Bonito, V.; Bouten, C.V.C.; Smits, A. Biomaterial-driven in situ cardiovascular tissue engineering-a multi-disciplinary perspective. NPJ Regen. Med. 2017, 2, 18. [Google Scholar] [CrossRef]
- Yacoub, M.H.; Tseng, Y.T.; Kluin, J.; Vis, A.; Stock, U.; Smail, H.; Sarathchandra, P.; Aikawa, E.; El-Nashar, H.; Chester, A.H.; et al. Valvulogenesis of a living, innervated pulmonary root induced by an acellular scaffold. Commun. Biol. 2023, 6, 1017. [Google Scholar] [CrossRef] [PubMed]
- Meccanici, F.; Notenboom, M.L.; Meijssen, J.; Smit, V.; van de Woestijne, P.C.; van den Bosch, A.E.; Helbing, W.A.; Bogers, A.; Takkenberg, J.J.M.; Roos-Hesselink, J.W. Long-term surgical outcomes of congenital supravalvular aortic stenosis: A systematic review, meta-analysis and microsimulation study. Eur. J. Cardiothorac. Surg. 2023, 65, ezad360. [Google Scholar] [CrossRef]
- Notenboom, M.L.; Rhellab, R.; Etnel, J.R.G.; van den Bogerd, N.; Veen, K.M.; Taverne, Y.; Helbing, W.A.; van de Woestijne, P.C.; Bogers, A.; Takkenberg, J.J.M. Aortic Valve Repair in Neonates, Infants and Children: A Systematic Review, Meta-Analysis and Microsimulation Study. Eur. J. Cardiothorac. Surg. 2023, 64, ezad284. [Google Scholar] [CrossRef] [PubMed]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Notenboom, M.L.; Van Hoof, L.; Schuermans, A.; Takkenberg, J.J.M.; Rega, F.R.; Taverne, Y.J.H.J. Aortic Valve Embryology, Mechanobiology, and Second Messenger Pathways: Implications for Clinical Practice. J. Cardiovasc. Dev. Dis. 2024, 11, 49. https://doi.org/10.3390/jcdd11020049
Notenboom ML, Van Hoof L, Schuermans A, Takkenberg JJM, Rega FR, Taverne YJHJ. Aortic Valve Embryology, Mechanobiology, and Second Messenger Pathways: Implications for Clinical Practice. Journal of Cardiovascular Development and Disease. 2024; 11(2):49. https://doi.org/10.3390/jcdd11020049
Chicago/Turabian StyleNotenboom, Maximiliaan L., Lucas Van Hoof, Art Schuermans, Johanna J. M. Takkenberg, Filip R. Rega, and Yannick J. H. J. Taverne. 2024. "Aortic Valve Embryology, Mechanobiology, and Second Messenger Pathways: Implications for Clinical Practice" Journal of Cardiovascular Development and Disease 11, no. 2: 49. https://doi.org/10.3390/jcdd11020049