
Comparative Analysis of Heart Regeneration: Searching for the Key to Heal the Heart—Part II: Molecular Mechanisms of Cardiac Regeneration
 , , and
, , and 
Abstract
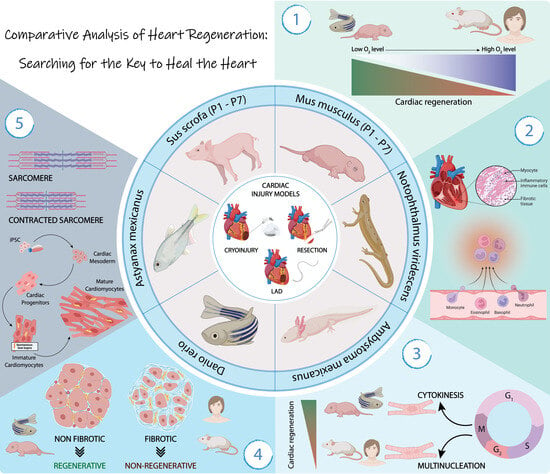
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Molecular Bases of Cardiac Regeneration
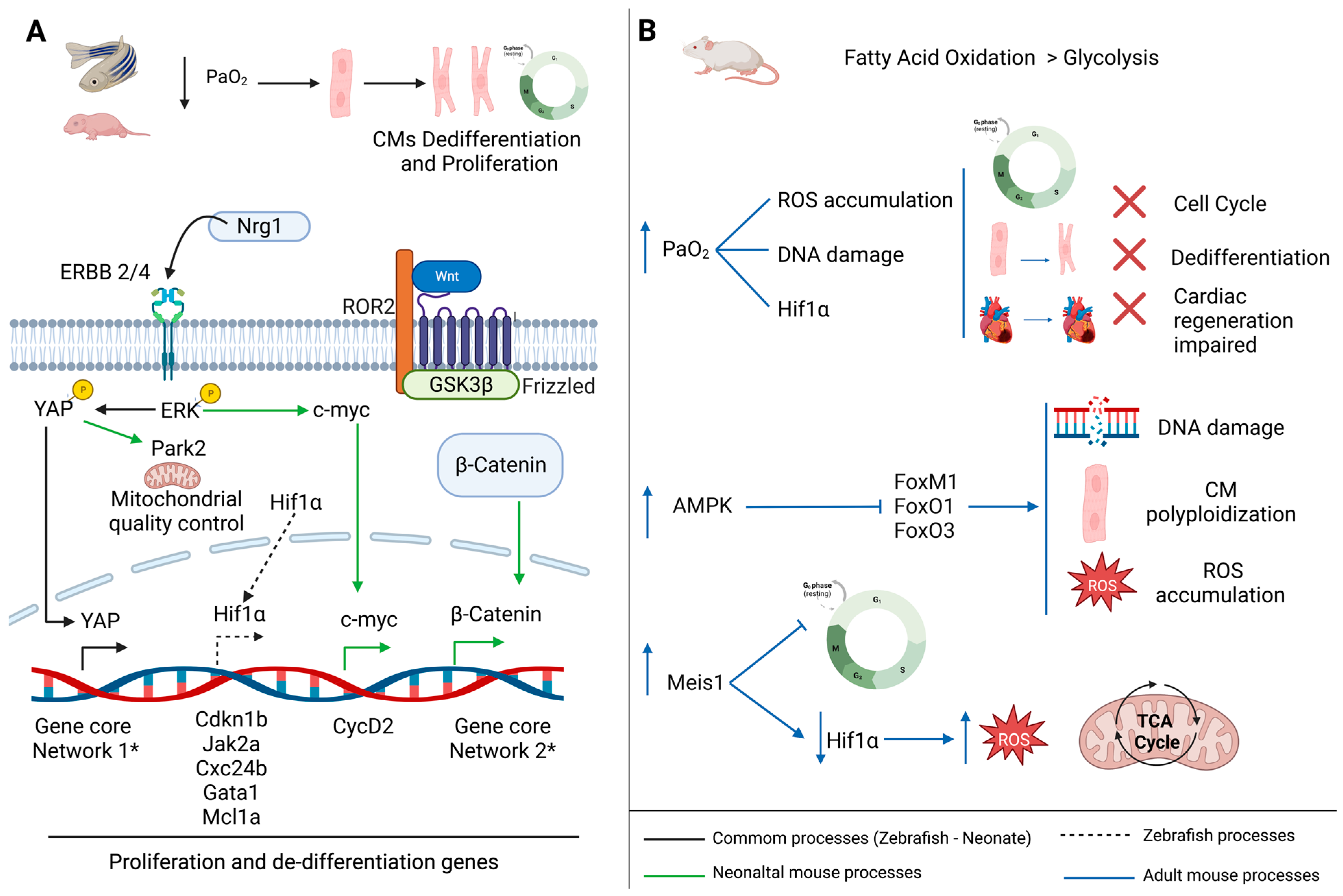
3. The Importance of a Depressed O2 Environment for Heart Regeneration
3.1. Hypoxic Environment Promotes Cardiomyocytes Proliferation
3.2. Metabolic Switch Promotes Cardiomyocytes Cell Cycle Exit
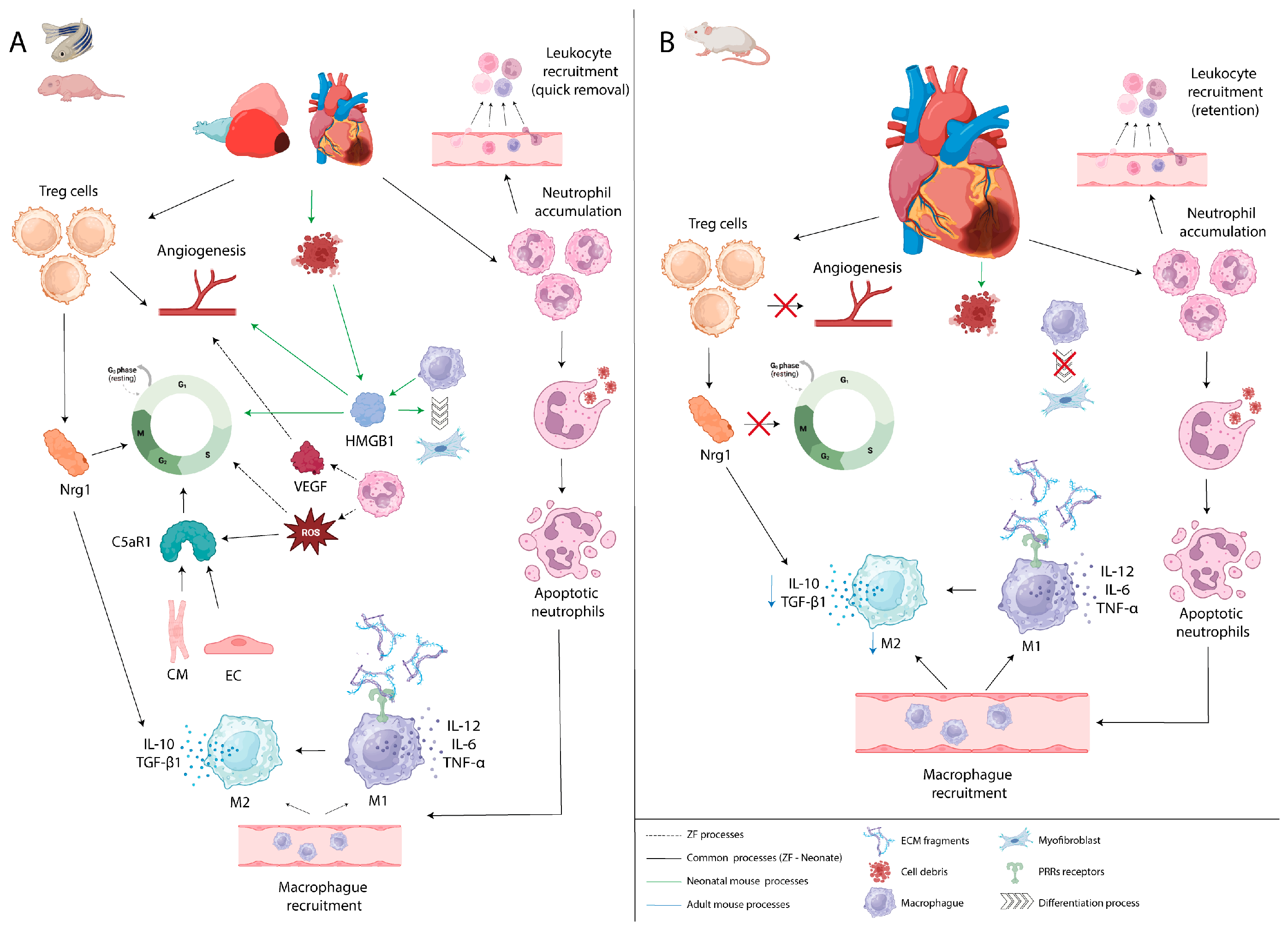
4. Inflammatory Process in Cardiac Regeneration
5. Reactivation of Cell Proliferation for Heart Repair
5.1. The Myocardial Inductors of Cardiomyocyte Proliferation
5.1.1. Cell Signaling
5.1.2. Transcription Factors
5.2. The Epicardial Inductors of Cardiomyocyte Proliferation
5.2.1. Cell Signaling
5.2.2. Transcription Factors
5.3. Other Mediators of Cardiomyocyte Proliferation
6. Modulation of Extracellular Matrix Deposition during Cardiac Repair
6.1. Zebrafish
6.2. Mice
7. The Role of Mechanical Stress in Cardiac Regeneration
8. Conclusions and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Khan, M.A.; Hashim, M.J.; Mustafa, H.; Baniyas, M.Y.; Al Suwaidi, S.K.B.M.; AlKatheeri, R.; Alblooshi, F.M.K.; Almatrooshi, M.E.A.H.; Alzaabi, M.E.H.; Al Darmaki, R.S.; et al. Global Epidemiology of Ischemic Heart Disease: Results from the Global Burden of Disease Study. Cureus 2020, 12, e9349. [Google Scholar] [CrossRef] [PubMed]
- Roth, G.A.; Mensah, G.A.; Johnson, C.O.; Addolorato, G.; Ammirati, E.; Baddour, L.M.; Barengo, N.C.; Beaton, A.; Benjamin, E.J.; Benziger, C.P.; et al. Global Burden of Cardiovascular Diseases and Risk Factors, 1990–2019: Update From the GBD 2019 Study. J. Am. Coll. Cardiol. 2020, 76, 2982–3021. [Google Scholar] [CrossRef] [PubMed]
- Thygesen, K.; Alpert, J.S.; Jaffe, A.S.; Chaitman, B.R.; Bax, J.J.; Morrow, D.A.; White, H.D. Fourth Universal Definition of Myocardial Infarction (2018). J. Am. Coll. Cardiol. 2018, 72, 2231–2264. [Google Scholar] [CrossRef] [PubMed]
- Lodrini, A.M.; Goumans, M.J. Cardiomyocytes Cellular Phenotypes After Myocardial Infarction. Front. Cardiovasc. Med. 2021, 8, 750510. [Google Scholar] [CrossRef]
- Jennings, R.; Sommers, H.; Smyth, G.; Flack, H.; Linn, H. Myocardial necrosis induced by temporary occlusion of a coronary artery in the dog. Arch. Pathol. 1960, 70, 68–78. [Google Scholar]
- Kathiresan, S.; Srivastava, D. Genetics of human cardiovascular disease. Cell 2012, 148, 1242–1257. [Google Scholar] [CrossRef] [PubMed]
- Lu, B.; Yu, H.; Zwartbol, M.; Ruifrok, W.P.; van Gilst, W.H.; de Boer, R.A.; Silljé, H.H.W. Identification of hypertrophy- and heart failure-associated genes by combining in vitro and in vivo models. Physiol. Genom. 2012, 44, 443–454. [Google Scholar] [CrossRef]
- Sarkar, K.; Cai, Z.; Gupta, R.; Parajuli, N.; Fox-Talbot, K.; Darshan, M.S.; Gonzalez, F.J.; Semenza, G.L. Hypoxia-inducible factor 1 transcriptional activity in endothelial cells is required for acute phase cardioprotection induced by ischemic preconditioning. Proc. Natl. Acad. Sci. USA 2012, 109, 10504–10509. [Google Scholar] [CrossRef]
- Bergmann, O.; Bhardwaj, R.D.; Bernard, S.; Zdunek, S.; Walsh, S.; Zupicich, J.; Alkass, K.; Buchholz, B.A.; Jovinge, S.; Frisén, J.; et al. Evidence for cardiomyocyte renewal in humans. Natl. Inst. Health 2009, 324, 98–102. [Google Scholar] [CrossRef]
- Senyo, S.E.; Steinhauser, M.L.; Pizzimenti, C.L.; Yang, V.K.; Cai, L.; Wang, M.; Wu, T.D.; Guerquin-Kern, J.L.; Lechene, C.P.; Lee, R.T. Mammalian heart renewal by pre-existing cardiomyocytes. Nature 2013, 493, 433–436. [Google Scholar] [CrossRef]
- Cassavaugh, J.; Lounsbury, K.M. Hypoxia-mediated biological control. J. Cell. Biochem. 2011, 112, 735–744. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L. Hypoxia-Inducible Factors in Physiology and Medicine. Cell 2012, 148, 399–408. [Google Scholar] [CrossRef] [PubMed]
- Santos, C.X.C.; Anilkumar, N.; Zhang, M.; Brewer, A.C.; Shah, A.M. Redox signaling in cardiac myocytes. Free. Radic. Biol. Med. 2011, 50, 777–793. [Google Scholar] [CrossRef] [PubMed]
- Puente, B.N.; Kimura, W.; Muralidhar, S.A.; Moon, J.; Amatruda, J.F.; Phelps, K.L.; Grinsfelder, D.; Rothermel, B.A.; Chen, R.; Garcia, J.A.; et al. The oxygen-rich postnatal environment induces cardiomyocyte cell-cycle arrest through DNA damage response. Cell 2014, 157, 565–579. [Google Scholar] [CrossRef] [PubMed]
- Kimura, W.; Xiao, F.; Canseco, D.C.; Muralidhar, S.; Thet, S.; Zhang, H.M.; Abderrahman, Y.; Chen, R.; Garcia, J.A.; Shelton, J.M.; et al. Hypoxia fate mapping identifies cycling cardiomyocytes in the adult heart. Nature 2015, 523, 226–230. [Google Scholar] [CrossRef]
- Nakada, Y.; Canseco, D.C.; Thet, S.; Abdisalaam, S.; Asaithamby, A.; Santos, C.X.; Shah, A.M.; Zhang, H.; Faber, J.E.; Kinter, M.T.; et al. Hypoxia induces heart regeneration in adult mice. Nature 2017, 541, 222–227. [Google Scholar] [CrossRef]
- Sakaguchi, A.; Nishiyama, C.; Kimura, W. Cardiac regeneration as an environmental adaptation. Biochim. Et Biophys. Acta-Mol. Cell Res. 2020, 1867, 118623. [Google Scholar] [CrossRef]
- Lawrence, J.; Xiao, D.L.; Xue, Q.; Rejali, M.; Yang, S.; Zhang, L. Prenatal nicotine exposure increases heart susceptibility to ischemia/reperfusion injury in adult offspring. J. Pharmacol. Exp. Ther. 2008, 324, 331–341. [Google Scholar] [CrossRef]
- Webster, W.S.; Abela, D. The effect of hypoxia in development. Birth Defects Res. Part C-Embryo Today Rev. 2007, 81, 215–228. [Google Scholar] [CrossRef]
- Cao, T.; Liccardo, D.; LaCanna, R.; Zhang, X.; Lu, R.; Finck, B.N.; Leigh, T.; Chen, X.; Drosatos, K.; Tian, Y. Fatty Acid Oxidation Promotes Cardiomyocyte Proliferation Rate but Does Not Change Cardiomyocyte Number in Infant Mice. Front. Cell Dev. Biol. 2019, 7, 42. [Google Scholar] [CrossRef]
- Lopaschuk, G.D.; Spafford, M.A.; Marsh, D.R. Glycolysis is predominant source of myocardial ATP production immediately after birth. Am. J. Physiol.-Heart Circ. Physiol. 1991, 261, H1698–705. [Google Scholar] [CrossRef] [PubMed]
- Kannan, S.; Kwon, C. Regulation of cardiomyocyte maturation during critical perinatal window. J. Physiol. 2020, 598, 2941–2956. [Google Scholar] [CrossRef] [PubMed]
- Lopaschuk, G.D.; Jaswal, J.S. Energy metabolic phenotype of the cardiomyocyte during development, differentiation, and postnatal maturation. J. Cardiovasc. Pharmacol. 2010, 56, 130–140. [Google Scholar] [CrossRef]
- Carley, A.N.; Taegtmeyer, H.; Lewandowski, E.D. Mechanisms Linking Energy Substrate Metabolism to the Function of the Heart. Physiol. Behav. 2014, 114, 717–729. [Google Scholar]
- Leu, M.; Ehler, E.; Perriard, J.C. Characterisation of postnatal growth of the murine heart. Anat. Embryol. 2001, 204, 217–224. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Liu, H.; Dudley, S.C. Reactive oxygen species originating from mitochondria regulate the cardiac sodium channel. Circ. Res. 2010, 107, 967–974. [Google Scholar] [CrossRef]
- Jopling, C.; Suñé, G.; Faucherre, A.; Fabregat, C.; Izpisua Belmonte, J.C. Hypoxia induces myocardial regeneration in zebrafish. Circulation 2012, 126, 3017–3027. [Google Scholar] [CrossRef]
- Marques, I.J.; Leito, J.T.D.; Spaink, H.P.; Testerink, J.; Jaspers, R.T.; Witte, F.; Van Den Berg, S.; Bagowski, C.P. Transcriptome analysis of the response to chronic constant hypoxia in zebrafish hearts. J. Comp. Physiol. B Biochem. Syst. Environ. Physiol. 2008, 178, 77–92. [Google Scholar] [CrossRef]
- Schioppa, T.; Uranchimeg, B.; Saccani, A.; Biswas, S.K.; Doni, A.; Rapisarda, A.; Bernasconi, S.; Saccani, S.; Nebuloni, M.; Vago, L.; et al. Regulation of the Chemokine Receptor CXCR4 by Hypoxia. J. Exp. Med. 2003, 198, 1391–1402. [Google Scholar] [CrossRef]
- Frolova, E.G.; Sopko, N.; Blech, L.; Popović, Z.B.; Li, J.; Vasanji, A.; Drumm, C.; Krukovets, I.; Jain, M.K.; Penn, M.S.; et al. Thrombospondin-4 regulates fibrosis and remodeling of the myocardium in response to pressure overload. FASEB J. 2012, 26, 2363–2373. [Google Scholar] [CrossRef]
- Cottage, C.T.; Bailey, B.; Fischer, K.M.; Avitable, D.; Collins, B.; Tuck, S.; Quijada, P.; Gude, N.; Alvarez, R.; Muraski, J.; et al. Cardiac progenitor cell cycling stimulated by Pim-1 kinase. Circ. Res. 2010, 106, 891–901. [Google Scholar] [CrossRef] [PubMed]
- Yoshioka, J.; Schulze, P.C.; Cupesi, M.; Sylvan, J.D.; MacGillivray, C.; Gannon, J.; Huang, H.; Lee, R.T. Thioredoxin-interacting protein controls cardiac hypertrophy through regulation of thioredoxin activity. Circulation 2004, 109, 2581–2586. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, B.V.; Kieller, D.M.; Quon, A.L.; Robertson, M.; Casey, J.R. Cardiac hypertrophy in anion exchanger 1-null mutant mice with severe hemolytic anemia. Am. J. Physiol.-Heart Circ. Physiol. 2007, 292, 1301–1312. [Google Scholar] [CrossRef] [PubMed]
- Penn, M.S.; Pastore, J.; Miller, T.; Aras, R. SDF-1 in myocardial repair. Gene Ther. 2012, 19, 583–587. [Google Scholar] [CrossRef]
- Chang, K.T.; Tsai, C.M.; Chiou, Y.C.; Chiu, C.H.; Jeng, K.S.; Huang, C.Y.F. IL-6 induces neuroendocrine dedifferentiation and cell proliferation in non-small cell lung cancer cells. Am. J. Physiol.-Lung Cell. Mol. Physiol. 2005, 289, 446–453. [Google Scholar] [CrossRef]
- Wehler, T.C.; Graf, C.; Biesterfeld, S.; Brenner, W.; Schadt, J.; Gockel, I.; Berger, M.R.; Thüroff, J.W.; Galle, P.R.; Moehler, M.; et al. Strong Expression of Chemokine Receptor CXCR 4 by Renal Cell Carcinoma Correlates with Advanced Disease. J. Oncol. 2008, 2008, 2–6. [Google Scholar] [CrossRef]
- Hashimoto, K.; Kodama, A.; Honda, T.; Hanashima, A.; Ujihara, Y.; Murayama, T.; Nishimatsu, S.I.; Mohri, S. Fam64a is a novel cell cycle promoter of hypoxic fetal cardiomyocytes in mice. Sci. Rep. 2017, 7, 4486. [Google Scholar] [CrossRef]
- Sun, Y.; Jiang, C.; Hong, H.; Liu, J.; Qiu, L.; Huang, Y.; Ye, L. Effects of hypoxia on cardiomyocyte proliferation and association with stage of development. Biomed. Pharmacother. 2019, 118, 109391. [Google Scholar] [CrossRef]
- Ye, L.; Qiu, L.; Feng, B.; Jiang, C.; Huang, Y.; Zhang, H.; Hong, H.; Liu, J. Role of Blood Oxygen Saturation During Post-Natal Human Cardiomyocyte Cell Cycle Activities. JACC Basic Transl. Sci. 2020, 5, 447–460. [Google Scholar] [CrossRef]
- Bolte, C.; Zhang, Y.; Wang, I.-C.; Kalin, T.V.; Molkentin, J.D.; Kalinichenko, V.V. Expression of Foxm1 transcription factor in cardiomyocytes is required for myocardial development. PLoS ONE 2011, 6, e22217. [Google Scholar] [CrossRef]
- Kalin, T.V.; Ustiyan, V.; Kalinichenko, V.V. Multiple faces of FoxM1 transcription factor: Lessons from transgenic mouse models. Cell Cycle 2011, 10, 396–405. [Google Scholar] [CrossRef] [PubMed]
- Evans-Anderson Heather, J.; Alfieri Christina, M.; Yutzey Katherine, E. Regulation of cardiomyocyte proliferation and myocardial growth during development by FOXO transcription factors. Circ. Res. 2008, 102, 686–694. [Google Scholar] [CrossRef] [PubMed]
- Ni, Y.G.; Berenji, K.; Wang, N.; Oh, M.; Sachan, N.; Dey, A.; Cheng, J.; Lu, G.; Morris, D.J.; Castrillon, D.H.; et al. Foxo transcription factors blunt cardiac hypertrophy by inhibiting calcineurin signaling. Circulation 2006, 114, 1159–1168. [Google Scholar] [CrossRef]
- Sengupta, A.; Kalinichenko, V.V.; Yutzey, K.E. FoxO and FoxM1 Transcription Factors Have Antagonistic Functions in Neonatal Cardiomyocyte Cell Cycle Withdrawal and IGF1 Gene Regulation. Criculation Res. 2013, 112, 267–277. [Google Scholar] [CrossRef] [PubMed]
- Mahmoud, A.I.; Kocabas, F.; Muralidhar, S.A.; Kimura, W.; Koura, A.S.; Thet, S.; Porrello, E.R.; Sadek, H.A. Meis1 regulates postnatal cardiomyocyte cell cycle arrest. Nature 2013, 497, 249–253. [Google Scholar] [CrossRef] [PubMed]
- Kocabas, F.; Zheng, J.; Thet, S.; Copeland, N.G.; Jenkins, N.A.; DeBerardinis, R.J.; Zhang, C.; Sadek, H.A. Meis1 regulates the metabolic phenotype and oxidant defense of hematopoietic stem cells. Blood 2012, 120, 4963–4972. [Google Scholar] [CrossRef]
- Koo, J.H.; Guan, K.L. Interplay between YAP/TAZ and Metabolism. Cell Metab. 2018, 28, 196–206. [Google Scholar] [CrossRef]
- von Gise, A.; Lin, Z.; Schlegelmilch, K.; Honor, L.B.; Pan, G.M.; Buck, J.N.; Ma, Q.; Ishiwata, T.; Zhou, B.; Camargo, F.D.; et al. YAP1, the nuclear target of Hippo signaling, stimulates heart growth through cardiomyocyte proliferation but not hypertrophy. Proc. Natl. Acad. Sci. USA 2012, 109, 2394–2399. [Google Scholar] [CrossRef]
- Leach, J.P.; Heallen, T.; Zhang, M.; Rahmani, M.; Morikawa, Y.; Hill, M.C.; Segura, A.; Willerson, J.T.; Martin, J.F. Hippo pathway deficiency reverses systolic heart failure after infarction. Nature 2017, 550, 260–264. [Google Scholar] [CrossRef]
- Tao, G.; Kahr, P.C.; Morikawa, Y.; Zhang, M.; Rahmani, M.; Heallen, T.R.; Li, L.; Sun, Z.; Olson, E.N.; Amendt, B.A.; et al. Pitx2 promotes heart repair by activating the antioxidant response after cardiac injury. Nature 2016, 534, 119–123. [Google Scholar] [CrossRef]
- Monroe, T.O.; Hill, M.C.; Morikawa, Y.; Leach, J.P.; Heallen, T.; Cao, S.; Krijger, P.H.; de Laat, W.; Wehrens, X.H.; Rodney, G.G.; et al. YAP Partially Reprograms Chromatin Accessibility to Directly Induce Adult Cardiogenesis In Vivo. Dev. Cell 2019, 48, 765–779.e7. [Google Scholar] [CrossRef]
- Li, L.; Tao, G.; Hill, M.C.; Zhang, M.; Morikawa, Y.; Martin, J.F. Pitx2 maintains mitochondrial function during regeneration to prevent myocardial fat deposition. Development 2018, 145, dev168609. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.; Liu, Z.; Desai, S.; Zhao, Y.; Liu, H.; Pannell, L.K.; Yi, H.; Wright, E.R.; Owen, L.B.; Dean-Colomb, W.; et al. Receptor tyrosine kinase ErbB2 translocates into mitochondria and regulates cellular metabolism. Nat. Commun. 2012, 3, 1271. [Google Scholar] [CrossRef] [PubMed]
- Gemberling, M.; Karra, R.; Dickson, A.L.; Poss, K.D. Nrg1 is an injury-induced cardiomyocyte mitogen for the endogenous heart regeneration program in zebrafish. eLife 2015, 4, e05871. [Google Scholar] [CrossRef]
- D’uva, G.; Aharonov, A.; Lauriola, M.; Kain, D.; Yahalom-Ronen, Y.; Carvalho, S.; Weisinger, K.; Bassat, E.; Rajchman, D.; Yifa, O.; et al. ERBB2 triggers mammalian heart regeneration by promoting cardiomyocyte dedifferentiation and proliferation. Nat. Cell Biol. 2015, 17, 627–638. [Google Scholar] [CrossRef] [PubMed]
- Aharonov, A.; Shakked, A.; Umansky, K.B.; Savidor, A.; Genzelinakh, A.; Kain, D.; Lendengolts, D.; Revach, O.-Y.; Morikawa, Y.; Dong, J.; et al. ERBB2 drives YAP activation and EMT-like processes during cardiac regeneration. Nat. Cell Biol. 2020, 22, 1346–1356. [Google Scholar] [CrossRef]
- Honkoop, H.; de Bakker, D.E.; Aharonov, A.; Kruse, F.; Shakked, A.; Nguyen, P.D.; de Heus, C.; Garric, L.; Muraro, M.J.; Shoffner, A.; et al. Single-cell analysis uncovers that metabolic reprogramming by ErbB2 signaling is essential for cardiomyocyte proliferation in the regenerating heart. eLife 2019, 8, e50163. [Google Scholar] [CrossRef]
- Ahuja, P.; Zhao, P.; Angelis, E.; Ruan, H.; Korge, P.; Olson, A.; Wang, Y.; Jin, E.S.; Jeffrey, F.M.; Portman, M.; et al. Myc controls transcriptional regulation of cardiac metabolism and mitochondrial biogenesis in response to pathological stress in mice. J. Clin. Investig. 2010, 120, 1494–1505. [Google Scholar] [CrossRef]
- Mills, R.J.; Titmarsh, D.M.; Koenig, X.; Parker, B.L.; Ryall, J.G.; Quaife-Ryan, G.A.; Voges, H.K.; Hodson, M.P.; Ferguson, C.; Drowley, L.; et al. Functional screening in human cardiac organoids reveals a metabolic mechanism for cardiomyocyte cell cycle arrest. Proc. Natl. Acad. Sci. USA 2017, 114, E8372–81. [Google Scholar] [CrossRef]
- Quaife-Ryan, G.A.; Mills, R.J.; Lavers, G.; Voges, H.K.; Vivien, C.J.; Elliott, D.A.; Ramialison, M.; Hudson, J.E.; Porrello, E.R. β-Catenin drives distinct transcriptional networks in proliferative and nonproliferative cardiomyocytes. Development 2020, 147, dev193417. [Google Scholar] [CrossRef]
- Murray, T.V.; Smyrnias, I.; Schnelle, M.; Mistry, R.K.; Zhang, M.; Beretta, M.; Martin, D.; Anilkumar, N.; de Silva, S.M.; Shah, A.M.; et al. Redox regulation of cardiomyocyte cell cycling via an ERK1/2 and c-Myc-dependent activation of cyclin D2 transcription. J. Mol. Cell. Cardiol. 2015, 79, 54–68. [Google Scholar] [CrossRef]
- Halder, G.; Johnson, R.L. Hippo signaling: Growth control and beyond. Development 2011, 138, 9–22. [Google Scholar] [CrossRef] [PubMed]
- Thompson, C.B. Wnt meets Warburg: Another piece in the puzzle? EMBO J. 2014, 33, 1420–1422. [Google Scholar] [CrossRef]
- Vallée, A.; Lecarpentier, Y.; Guillevin, R.; Vallée, J.N. Aerobic Glycolysis Hypothesis Through WNT/Beta-Catenin Pathway in Exudative Age-Related Macular Degeneration. J. Mol. Neurosci. 2017, 62, 368–379. [Google Scholar] [CrossRef] [PubMed]
- Vallée, A.; Vallée, J.N. Warburg effect hypothesis in autism Spectrum disorders. Mol. Brain 2018, 11, 1. [Google Scholar] [CrossRef] [PubMed]
- Beltrami, A.P.; Urbanek, K.; Kajstura, J.; Yan, S.-M.; Finato, N.; Bussani, R.; Nadal-Ginard, B.; Silvestri, F.; Leri, A.; Beltrami, C.A.; et al. Evidence that human cardiac myocytes divide after myocardial infarction. N. Engl. J. Med. 2001, 344, 1750–1757. [Google Scholar] [CrossRef]
- Jonker, S.S.; Louey, S.; Giraud, G.D.; Thornburg, K.L.; Faber, J.J. Timing of cardiomyocyte growth, maturation, and attrition in perinatal sheep. FASEB J. 2015, 29, 4346–4357. [Google Scholar] [CrossRef]
- Hirose, K.; Payumo, A.Y.; Cutie, S.; Hoang, A.; Zhang, H.; Guyot, R.; Lunn, D.; Bigley, R.B.; Yu, H.; Wang, J.; et al. Evidence for hormonal control of heart regenerative capacity during endothermy acquisition. Science 2019, 364, 184–188. [Google Scholar] [CrossRef] [PubMed]
- Makarieva, A.M.; Gorshkov, V.G.; Li, B.L.; Chown, S.L.; Reich, P.B.; Gavrilov, V.M. Mean mass-specific metabolic rates are strikingly similar across life’s major domains: Evidence for life’s metabolic optimum. Proc. Natl. Acad. Sci. USA 2008, 105, 16994–16999. [Google Scholar] [CrossRef]
- Aurora, A.B.; Olson, E.N. Immune modulation of stem cells and regeneration. Cell Stem Cell 2014, 15, 14–25. [Google Scholar] [CrossRef]
- Godwin, J.W.; Brockes, J.P. Regeneration, tissue injury and the immune response. J. Anat. 2006, 209, 423–432. [Google Scholar] [CrossRef] [PubMed]
- Aurora, A.B.; Porrello, E.R.; Tan, W.; Mahmoud, A.I.; Hill, J.A.; Bassel-Duby, R.; Sadek, H.A.; Olson, E.N. Macrophages are required for neonatal heart regeneration. J. Clin. Investig. 2014, 124, 1382–1392. [Google Scholar] [CrossRef] [PubMed]
- Lavine, K.J.; Epelman, S.; Uchida, K.; Weber, K.J.; Nichols, C.G.; Schilling, J.D.; Ornitz, D.M.; Randolph, G.J.; Mann, D.L. Distinct macrophage lineages contribute to disparate patterns of cardiac recovery and remodeling in the neonatal and adult heart. Proc. Natl. Acad. Sci. USA 2014, 111, 16029–16034. [Google Scholar] [CrossRef] [PubMed]
- Lai, S.L.; Marín-Juez, R.; Moura, P.L.; Kuenne, C.; Lai, J.K.H.; Tsedeke, A.T.; Guenther, S.; Looso, M.; Stainier, D.Y.R. Reciprocal analyses in zebrafish and medaka reveal that harnessing the immune response promotes cardiac regeneration. eLife 2017, 6, e25605. [Google Scholar] [CrossRef]
- Mescher, A.L.; Neff, A.W. Regenerative capacity and the developing immune system. Adv. Biochem. Eng. Biotechnol. 2005, 93, 39–66. [Google Scholar]
- Bianchi, M.E.; Crippa, M.P.; Manfredi, A.A.; Mezzapelle, R.; Rovere Querini, P.; Venereau, E. High-mobility group box 1 protein orchestrates responses to tissue damage via inflammation, innate and adaptive immunity, and tissue repair. Immunol. Rev. 2017, 280, 74–82. [Google Scholar] [CrossRef]
- Klune, J.R.; Dhupar, R.; Cardinal, J.; Billiar, T.R.; Tsung, A. HMGB1: Endogenous danger signaling. Mol. Med. 2008, 14, 476–484. [Google Scholar] [CrossRef]
- Andrassy, M.; Volz, H.; Igwe, J.; Funke, B.; Eichberger, S.; Kaya, Z.; Buss, S.; Autschbach, F.; Pleger, S.; Lukic, I.; et al. High-mobility group box-1 in ischemia-reperfusion injury of the heart. Circulation 2008, 117, 3216–3226. [Google Scholar] [CrossRef]
- Omiya, S.; Omori, Y.; Taneike, M.; Protti, A.; Yamaguchi, O.; Akira, S.; Shah, A.M.; Nishida, K.; Otsu, K. Toll-like receptor 9 prevents cardiac rupture after myocardial infarction in mice independently of inflammation. Am. J. Physiol. Heart Circ. Physiol. 2016, 311, H1485–H1497. [Google Scholar] [CrossRef]
- Ivanov, S.; Dragoi, A.M.; Wang, X.; Dallacosta, C.; Louten, J.; Musco, G.; Sitia, G.; Yap, G.S.; Wan, Y.; Biron, C.A.; et al. A novel role for HMGB1 in TLR9-mediated inflammatory responses to CpG-DNA. Blood 2007, 110, 1970–1981. [Google Scholar] [CrossRef]
- Chen, G.; Nuñez, G. Sterile inflammation: Sensing and reacting to damage. Nat. Rev. Immunol. 2010, 10, 826–837. [Google Scholar] [CrossRef]
- Timmers, L.; Pasterkamp, G.; Hoog, V.C.; Arslan, F.; Appelman, Y.; Kleijn, D.P. The innate immune response in reperfused myocardium. Cardiovasc. Res. 2012, 94, 276–283. [Google Scholar] [CrossRef] [PubMed]
- Nacu, E.; Glausch, M.; Le, H.Q.; Damanik, F.F.; Schuez, M.; Knapp, D.; Khattak, S.; Richter, T.; Tanaka, E.M. Connective tissue cells, but not muscle cells, are involved in establishing the proximo-distal outcome of limb regeneration in the axolotl. Development 2013, 140, 513–518. [Google Scholar] [CrossRef] [PubMed]
- Yanai, H.; Ban, T.; Wang, Z. HMGB proteins function as universal sentinels for nucleic-acid-mediated innate immune responses. Nature 2009, 462, 99–103. [Google Scholar] [CrossRef] [PubMed]
- Benias, P.C.; Wells, R.G.; Sackey-Aboagye, B. Structure and Distribution of an Unrecognized Interstitium in Human Tissues. Sci. Rep. 2018, 8, 4947. [Google Scholar] [CrossRef] [PubMed]
- Steinman, R.M.; Cohn, Z.A. Identification of a novel cell type in peripheral lymphoid organs of mice. I. Morphology, quantitation, tissue distribution. J. Exp. Med. 1973, 137, 1142–1162. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, C. Role of dendritic cells in cardiovascular diseases. World J. Cardiol. 2010, 2, 357–364. [Google Scholar] [CrossRef]
- Zhang, J.; Yu, Z.X.; Fujita, S.; Yamaguchi, M.L.; Ferrans, V.J. Interstitial dendritic cells of the rat heart. Quantitative and ultrastructural changes in experimental myocardial infarction. Circulation 1993, 87, 909–920. [Google Scholar] [CrossRef]
- Gallego-Colon, E.; Sampson, R.D.; Sattler, S.; Schneider, M.D.; Rosenthal, N.; Tonkin, J. Cardiac-Restricted IGF-1Ea Overexpression Reduces the Early Accumulation of Inflammatory Myeloid Cells and Mediates Expression of Extracellular Matrix Remodelling Genes after Myocardial Infarction. Mediators Inflamm. 2015, 2015, 484357. [Google Scholar] [CrossRef]
- Horckmans, M.; Ring, L.; Duchene, J.; Santovito, D.; Schloss, M.J.; Drechsler, M.; Weber, C.; Soehnlein, O.; Steffens, S. Neutrophils orchestrate post-myocardial infarction healing by polarizing macrophages towards a reparative phenotype. Eur. Heart J. 2017, 38, 187–197. [Google Scholar] [CrossRef]
- Summers, C.; Rankin, S.M.; Condliffe, A.M.; Singh, N.; Peters, A.M.; Chilvers, E.R. Neutrophil kinetics in health and disease. Trends Immunol. 2010, 31, 318–324. [Google Scholar] [CrossRef]
- Gong, Y.; Koh, D.R. Neutrophils promote inflammatory angiogenesis via release of preformed VEGF in an in vivo corneal model. Cell Tissue Res. 2010, 339, 437–448. [Google Scholar] [CrossRef] [PubMed]
- Marín-Juez, R.; Marass, M.; Gauvrit, S.; Rossi, A.; Lai, S.L.; Materna, S.C.; Black, B.L.; Stainier, D.Y.R. Fast revascularization of the injured area is essential to support zebrafish heart regeneration. Proc. Natl. Acad. Sci. USA 2016, 113, 11237–11242. [Google Scholar] [CrossRef] [PubMed]
- Frangogiannis, N.G. The role of transforming growth factor (TGF)-β in the infarcted myocardium. J. Thorac. Dis. 2017, 9 (Suppl. S1), S52–S63. [Google Scholar] [CrossRef]
- Sattler, S.; Rosenthal, N. The neonate versus adult mammalian immune system in cardiac repair and regeneration. Biochim. Biophys. Acta 2016, 1863, 1813–1821. [Google Scholar] [CrossRef] [PubMed]
- Lörchner, H.; Pöling, J.; Gajawada, P.; Hou, Y.; Polyakova, V.; Kostin, S.; Adrian-Segarra, J.M.; Boettger, T.; Wietelmann, A.; Warnecke, H.; et al. Myocardial healing requires Reg3β-dependent accumulation of macrophages in the ischemic heart. Nat. Med. 2015, 21, 353–362. [Google Scholar] [CrossRef] [PubMed]
- Ravichandran, K.S. Beginnings of a good apoptotic meal: The find-me and eat-me signaling pathways. Immunity 2011, 35, 445–455. [Google Scholar] [CrossRef]
- Elliott, M.R.; Koster, K.M.; Murphy, P.S. Efferocytosis Signaling in the Regulation of Macrophage Inflammatory Responses. J. Immunol. 2017, 198, 1387–1394. [Google Scholar] [CrossRef]
- Ortega-Gómez, A.; Perretti, M.; Soehnlein, O. Resolution of inflammation: An integrated view. EMBO Mol. Med. 2013, 5, 661–674. [Google Scholar] [CrossRef]
- Natarajan, N.; Abbas, Y.; Bryant, D.M.; Gonzalez-Rosa, J.M.; Sharpe, M.; Uygur, A.; Cocco-Delgado, L.H.; Ho, N.N.; Gerard, N.P.; Gerard, C.J.; et al. Complement receptor C5AR1 plays an evolutionarily conserved role in successful cardiac regeneration. Circulation 2018, 137, 2152–2165. [Google Scholar] [CrossRef]
- Kawaguchi, M.; Takahashi, M.; Hata, T.; Kashima, Y.; Usui, F.; Morimoto, H.; Izawa, A.; Takahashi, Y.; Masumoto, J.; Koyama, J.; et al. Inflammasome activation of cardiac fibroblasts is essential for myocardial ischemia/reperfusion injury. Circulation 2011, 123, 594–604. [Google Scholar] [CrossRef] [PubMed]
- Niethammer, P.; Grabher, C.; Look, A.T.; Mitchison, T.J. A tissue-scale gradient of hydrogen peroxide mediates rapid wound detection in zebrafish. Nature 2009, 459, 996–999. [Google Scholar] [CrossRef] [PubMed]
- Muntean, D.M.; Sturza, A.; Dănilă, M.D.; Borza, C.; Duicu, O.M.; Mornoș, C. The Role of Mitochondrial Reactive Oxygen Species in Cardiovascular Injury and Protective Strategies. Oxid. Med. Cell Longev. 2016, 2016, 8254942. [Google Scholar] [CrossRef] [PubMed]
- Frangogiannis, N.G. Regulation of the inflammatory response in cardiac repair. Circ. Res. 2012, 110, 159–173. [Google Scholar] [CrossRef] [PubMed]
- Corbett, S.A.; Schwarzbauer, J.E. Fibronectin-fibrin cross-linking: A regulator of cell behavior. Trends Cardiovasc. Med. 1998, 8, 357–362. [Google Scholar] [CrossRef]
- Flick, M.J.; Du, X.; Witte, D.P.; Jirousková, M.; Soloviev, D.A.; Busuttil, S.J.; Plow, E.F.; Degen, J.L. Leukocyte engagement of fibrin(ogen) via the integrin receptor alphaMbeta2/Mac-1 is critical for host inflammatory response in vivo. J. Clin. Investig. 2004, 113, 1596–1606. [Google Scholar] [CrossRef]
- Smiley, S.T.; King, J.A.; Hancock, W.W. Fibrinogen stimulates macrophage chemokine secretion through toll-like receptor 4. J. Immunol. 2001, 167, 2887–2894. [Google Scholar] [CrossRef]
- Oyama, J.I.; Blais, C., Jr.; Liu, X.; Pu, M.; Kobzik, L.; Kelly, R.A.; Bourcier, T. Reduced myocardial ischemia-reperfusion injury in toll-like receptor 4-deficient mice. Circulation 2004, 109, 784–789. [Google Scholar] [CrossRef]
- Arslan, F.; Smeets, M.B.; O’Neill, L.A.; Keogh, B.; McGuirk, P.; Timmers, L.; Tersteeg, C.; Hoefer, I.E.; Doevendans, P.A.; Pasterkamp, G.; et al. Myocardial ischemia/reperfusion injury is mediated by leukocytic toll-like receptor-2 and reduced by systemic administration of a novel anti-toll-like receptor-2 antibody. Circulation 2010, 121, 80–90. [Google Scholar] [CrossRef]
- de Preux Charles, A.S.; Bise, T.; Baier, F.; Sallin, P.; Jaźwińska, A. Preconditioning boosts regenerative programmes in the adult zebrafish heart. Open Biol. 2016, 6, 160101. [Google Scholar] [CrossRef]
- Han, C.; Nie, Y.; Lian, H.; Liu, R.; He, F.; Huang, H.; Hu, S. Acute inflammation stimulates a regenerative response in the neonatal mouse heart. Cell Res. 2015, 25, 1137–1151. [Google Scholar] [CrossRef]
- Frangogiannis, N.G. The inflammatory response in myocardial injury, repair, and remodelling. Nat. Rev. Cardiol. 2014, 11, 255–265. [Google Scholar] [CrossRef]
- González-Rosa, J.M.; Martín, V.; Peralta, M.; Torres, M.; Mercader, N. Extensive scar formation and regression during heart regeneration after cryoinjury in zebrafish. Development 2011, 138, 1663–1674. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Iranzo, H.; Galardi-Castilla, M.; Sanz-Morejón, A.; González-Rosa, J.M.; Costa, R.; Ernst, A.; de Aja, J.S.; Langa, X.; Mercader, N. Transient fibrosis resolves via fibroblast inactivation in the regenerating zebrafish heart. Proc. Natl. Acad. Sci. USA 2018, 115, 4188–4193. [Google Scholar] [CrossRef] [PubMed]
- Shinde, A.V.; Frangogiannis, N.G. Fibroblasts in myocardial infarction: A role in inflammation and repair. J. Mol. Cell. Cardiol. 2014, 70, 74–82. [Google Scholar] [CrossRef] [PubMed]
- Weirather, J.; Hofmann, U.D.; Beyersdorf, N.; Ramos, G.C.; Vogel, B.; Frey, A.; Ertl, G.; Kerkau, T.; Frantz, S. Foxp3+ CD4+ T cells improve healing after myocardial infarction by modulating monocyte/macrophage differentiation. Circ. Res. 2014, 115, 55–67. [Google Scholar] [CrossRef] [PubMed]
- Zouggari, Y.; Ait-Oufella, H.; Waeckel, L.; Vilar, J.; Loinard, C.; Cochain, C.; Récalde, A.; Duriez, M.; Levy, B.I.; Lutgens, E.; et al. Regulatory T cells modulate postischemic neovascularization. Circulation 2009, 120, 1415–1425. [Google Scholar] [CrossRef]
- Hui, S.P.; Sheng, D.Z.; Sugimoto, K.; Gonzalez-Rajal, A.; Nakagawa, S.; Hesselson, D.; Kikuchi, K. Zebrafish Regulatory T Cells Mediate Organ-Specific Regenerative Programs. Dev. Cell 2017, 43, 659–672.e5. [Google Scholar] [CrossRef]
- Zacchigna, S.; Martinelli, V.; Moimas, S.; Colliva, A.; Anzini, M.; Nordio, A.; Costa, A.; Rehman, M.; Vodret, S.; Pierro, C.; et al. Paracrine effect of regulatory T cells promotes cardiomyocyte proliferation during pregnancy and after myocardial infarction. Nat. Commun. 2018, 9, 2432. [Google Scholar] [CrossRef]
- Godwin, J.W.; Debuque, R.; Salimova, E.; Rosenthal, N.A. Heart regeneration in the salamander relies on macrophage-mediated control of fibroblast activation and the extracellular landscape. NPJ Regen. Med. 2017, 2, 22. [Google Scholar] [CrossRef]
- Peterson, E.A.; Sun, J.; Wang, J. Leukocyte-Mediated Cardiac Repair after Myocardial Infarction in Non-Regenerative vs. Regenerative Systems. J. Cardiovasc. Dev. Dis. 2022, 9, 63. [Google Scholar] [CrossRef] [PubMed]
- Saxena, A.; Chen, W.; Su, Y.; Rai, V.; Uche, O.U.; Li, N.; Frangogiannis, N.G. IL-1 Induces Proinflammatory Leukocyte Infiltration and Regulates Fibroblast Phenotype in the Infarcted Myocardium. J. Immunol. 2013, 191, 4838–4848. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Chen, X.; Cao, W. Plasticity of mesenchymal stem cells in immunomodulation: Pathological and therapeutic implications. Nat. Immunol. 2014, 15, 1009–1016. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, J.M.; Youd, M.E.; Lodie, T.A. Immunomodulatory activity of mesenchymal stem cells. Curr. Stem Cell Res. Ther. 2011, 6, 297–316. [Google Scholar] [CrossRef]
- Frangogiannis, N.G.; Mendoza, L.H.; Lindsey, M.L.; Ballantyne, C.M.; Michael, L.H.; Smith, C.W.; Entman, M.L. IL-10 is induced in the reperfused myocardium and may modulate the reaction to injury. J. Immunol. 2000, 165, 2798–2808. [Google Scholar] [CrossRef]
- Saxena, A.; Dobaczewski, M.; Rai, V.; Haque, Z.; Chen, W.; Li, N.; Frangogiannis, N.G. Regulatory T cells are recruited in the infarcted mouse myocardium and may modulate fibroblast phenotype and function. Am. J. Physiol. Heart Circ. Physiol. 2014, 307, H1233–H1242. [Google Scholar] [CrossRef]
- Kehat, I.; Molkentin, J.D. Molecular pathways underlying cardiac remodeling during pathophysiological stimulation. Circulation 2010, 122, 2727–2735. [Google Scholar] [CrossRef]
- Zhou, B.; Lin, Z.; Pu, W.T.; Zhou, B.; Lin, Z.; Pu, W.T. Mammalian Myocardial Regeneration. In Muscle: Fundamental Biology and Mechanisms of Disease; Academic Press: Cambridge, MA, USA, 2012; pp. 555–569. [Google Scholar]
- Lin, Z.; Pu, W.T. Harnessing Hippo in the heart: Hippo/Yap signaling and applications to heart regeneration and rejuvenation. Stem Cell Res. 2014, 13, 571–581. [Google Scholar] [CrossRef]
- Boopathy, G.T.K.; Hong, W. Role of Hippo Pathway-YAP/TAZ Signaling in Angiogenesis. Front. Cell Dev. Biol. 2019, 7, 49. [Google Scholar] [CrossRef]
- Baliga, R.; Pimental, D.; Zhao, Y.; Simmons, W.; Marchionni, M.; Sawyer, D.; Kelly, R. NRG-1-induced cardiomyocyte hypertrophy. Role of PI-3-kinase, p70(S6K), and MEK-MAPK-RSK. Am. J. Physiol.-Heart Circ. Physiol. 1999, 277, H2026–H2037. [Google Scholar] [CrossRef]
- Bersell, K.; Arab, S.; Haring, B.; Kühn, B. Neuregulin1/ErbB4 Signaling Induces Cardiomyocyte Proliferation and Repair of Heart Injury. Cell 2009, 138, 257–270. [Google Scholar] [CrossRef]
- Engel, F.B.; Schebesta, M.; Duong, M.; Lu, G.; Ren, S.; Madwed, J.; Jiang, H.; Wang, Y.; Keating, M. p38 MAP kinase inhibition enables proliferation of adult mammalian cardiomyocytes. Genes. Dev. 2005, 19, 1175–1187. [Google Scholar] [CrossRef] [PubMed]
- Han, Z.; Wang, X.; Xu, Z.; Cao, Y.; Gong, R.; Yu, Y.; Yu, Y.; Guo, X.; Liu, S.; Yu, M.; et al. ALKBH5 regulates cardiomyocyte proliferation and heart regeneration by demethylating the mRNA of YTHDF1. Theranostics 2021, 11, 3000–3016. [Google Scholar] [CrossRef] [PubMed]
- Lan, C.; Cao, N.; Chen, C.; Qu, S.; Fan, C.; Luo, H.; Zeng, A.; Yu, C.; Xue, Y.; Ren, H.; et al. Progesterone, via yes-associated protein, promotes cardiomyocyte proliferation and cardiac repair. Cell Prolif. 2020, 53, e12910. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Feng, J.; Song, S.; Li, H.; Yang, H.; Zhou, B.; Li, Y.; Yue, Z.; Lian, H.; Liu, L.; et al. gp130 Controls Cardiomyocyte Proliferation and Heart Regeneration. Circulation 2020, 142, 967–982. [Google Scholar] [CrossRef]
- Qu, S.; Liao, Q.; Yu, C.; Chen, Y.; Luo, H.; Xia, X.; He, D.; Xu, Z.; Jose, P.A.; Li, Z.; et al. LKB1 suppression promotes cardiomyocyte regeneration via LKB1-AMPK-YAP axis. Bosn. J. Basic Med. Sci. 2022, 22, 772–783. [Google Scholar] [CrossRef]
- Ganapathy, B.; Nandhagopal, N.; Polizzotti, B.D.; Bennett, D.; Asan, A.; Wu, Y.; Kühn, B. Neuregulin-1 Administration Protocols Sufficient for Stimulating Cardiac Regeneration in Young Mice Do Not Induce Somatic, Organ, or Neoplastic Growth. PLoS ONE 2016, 11, e0155456. [Google Scholar] [CrossRef]
- Formiga, F.R.; Pelacho, B.; Garbayo, E.; Imbuluzqueta, I.; Díaz-herráez, P.; Abizanda, G.; Gavira, J.J.; Simón-yarza, T.; Albiasu, E.; Tamayo, E.; et al. Controlled delivery of fibroblast growth factor-1 and neuregulin-1 from biodegradable microparticles promotes cardiac repair in a rat myocardial infarction model through activation of endogenous regeneration. J. Control. Release 2014, 173, 132–139. [Google Scholar] [CrossRef]
- Engel, F.B.; Hsieh, P.C.H.; Lee, R.T.; Keating, M.T. FGF1/p38 MAP kinase inhibitor therapy induces cardiomyocyte mitosis, reduces scarring, and rescues function after myocardial infarction. Proc. Natl. Acad. Sci. USA 2006, 103, 15546–15551. [Google Scholar] [CrossRef]
- Shoffner, A.; Cigliola, V.; Lee, N.; Ou, J.; Poss, K.D. Tp53 Suppression Promotes Cardiomyocyte Proliferation during Zebrafish Heart Regeneration. Cell Rep. 2020, 32, 108089. [Google Scholar] [CrossRef]
- Arora, H.; Lavin, A.C.; Balkan, W.; Hare, J.M.; White, A. Neuregulin-1, in a Conducive Milieu with Wnt/BMP/Retinoic Acid, Prolongs the Epicardial-Mediated Cardiac Regeneration Capacity of Neonatal Heart Explants. J. Stem Cells Regen. Med. 2021, 17, 18–27. [Google Scholar] [PubMed]
- Missinato, M.A.; Saydmohammed, M.; Zuppo, D.A.; Rao, K.S.; Opie, G.W.; Kühn, B.; Tsang, M. Dusp6 attenuates Ras/MAPK signaling to limit zebrafish heart regeneration. Development 2018, 145, 157206. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.P.; Umbarkar, P.; Guo, Y.; Force, T.; Gupte, M.; Lal, H. Inhibition of GSK-3 to induce cardiomyocyte proliferation: A recipe for in situ cardiac regeneration. Cardiovasc. Res. 2019, 115, 20–30. [Google Scholar] [CrossRef] [PubMed]
- Yusuf, A.M.; Qaisar, R.; Al-Tamimi, A.O.; Jayakumar, M.N.; Woodgett, J.R.; Koch, W.J.; Ahmad, F. Cardiomyocyte-GSK-3β deficiency induces cardiac progenitor cell proliferation in the ischemic heart through paracrine mechanisms. J. Cell. Physiol. 2022, 237, 1804–1817. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Xu, T.; Li, M.; Li, C.; Ma, Y.; Chen, G.; Sun, Y.; Zheng, H.; Wu, G.; Liao, W.; et al. Inhibition of SENP2-mediated Akt deSUMOylation promotes cardiac regeneration via activating Akt pathway. Clin. Sci. 2021, 135, 811–828. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Yang, T.; Wei, T.; Zhou, L.; Shan, T.; Chen, J.; Gu, L.; Chen, B.; Liu, L.; Jiang, Q.; et al. CDK9 binds and activates SGK3 to promote cardiac repair after injury via the GSK-3β/β-catenin pathway. Front. Cardiovasc. Med. 2022, 9, 970745. [Google Scholar] [CrossRef]
- Li, Y.; Wei, T.; Fan, Y.; Shan, T.; Sun, J.; Chen, B.; Wang, Z.; Gu, L.; Yang, T.; Liu, L.; et al. Serine/Threonine-Protein Kinase 3 Facilitates Myocardial Repair After Cardiac Injury Possibly Through the Glycogen Synthase Kinase-3β/β-Catenin Pathway. J. Am. Heart Assoc. 2021, 10, e022802. [Google Scholar] [CrossRef]
- Magadum, A.; Singh, N.; Kurian, A.A.; Munir, I.; Mehmood, T.; Brown, K.; Sharkar, M.T.K.; Chepurko, E.; Sassi, Y.; Oh, J.G.; et al. Pkm2 Regulates Cardiomyocyte Cell Cycle and Promotes Cardiac Regeneration. Circulation 2020, 141, 1249–1265. [Google Scholar] [CrossRef]
- Alam, P.; Haile, B.; Arif, M.; Pandey, R.; Rokvic, M.; Nieman, M.; Maliken, B.D.; Paul, A.; Wang, Y.; Sadayappan, S.; et al. Inhibition of Senescence-Associated Genes Rb1 and Meis2 in Adult Cardiomyocytes Results in Cell Cycle Reentry and Cardiac Repair Post–Myocardial Infarction. J. Am. Heart Assoc. 2019, 8, e012089. [Google Scholar] [CrossRef]
- Aksoz, M.; Turan, R.D.; Albayrak, E.; Kocabas, F. Emerging Roles of Meis1 in Cardiac Regeneration, Stem Cells and Cancer. Curr. Drug Targets 2018, 19, 181–190. [Google Scholar] [CrossRef]
- Hubert, F.; Payan, S.M.; Pelce, E.; Bouchard, L.; Sturny, R.; Lenfant, N.; Mottola, G.; Collart, F.; Kelly, R.G.; Rochais, F. FGF10 promotes cardiac repair through a dual cellular mechanism increasing cardiomyocyte renewal and inhibiting fibrosis. Cardiovasc. Res. 2022, 118, 2625–2637. [Google Scholar] [CrossRef] [PubMed]
- Yu, W.; Huang, X.; Tian, X.; Zhang, H.; He, L.; Wang, Y.; Nie, Y.; Hu, S.; Lin, Z.; Zhou, B.; et al. GATA4 regulates Fgf16 to promote heart repair after injury. Development 2016, 143, 936–949. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.; Poss, K.D. The epicardium as a hub for heart regeneration. Nat. Rev. Cardiol. 2018, 15, 631–647. [Google Scholar] [CrossRef] [PubMed]
- Magadum, A.; Singh, N.; Kurian, A.A.; Sharkar, M.T.K.; Chepurko, E.; Zangi, L. Ablation of a Single N-Glycosylation Site in Human FSTL 1 Induces Cardiomyocyte Proliferation and Cardiac Regeneration. Mol. Ther.-Nucleic Acids 2018, 13, 133–143. [Google Scholar] [CrossRef]
- Peters, M.C.; Di Martino, S.; Boelens, T.; Qin, J.; Van Mil, A.; Doevendans, P.A.; Chamuleau, S.A.J.; Sluijter, J.P.G.; Neef, K. Follistatin-like 1 promotes proliferation of matured human hypoxic iPSC-cardiomyocytes and is secreted by cardiac fibroblasts. Mol. Ther.-Methods Clin. Dev. 2022, 25, 3–16. [Google Scholar] [CrossRef]
- Sun, J.; Peterson, E.A.; Wang, A.Z.; Ou, J.; Smith, K.E.; Poss, K.D.; Wang, J. hapln1 Defines an Epicardial Cell Subpopulation Required for Cardiomyocyte Expansion During Heart Morphogenesis and Regeneration. Circulation 2022, 146, 48–63. [Google Scholar] [CrossRef]
- Wasserman, A.H.; Huang, A.R.; Lewis-Israeli, Y.R.; Dooley, M.D.; Mitchell, A.L.; Venkatesan, M.; Aguirre, A. Oxytocin promotes epicardial cell activation and heart regeneration after cardiac injury. Front. Cell Dev. Biol. 2022, 10, 985298. [Google Scholar] [CrossRef]
- El-Sammak, H.; Yang, B.; Guenther, S.; Chen, W.; Marín-Juez, R.; Stainier, D.Y.R. A Vegfc-Emilin2a-Cxcl8a Signaling Axis Required for Zebrafish Cardiac Regeneration. Circ. Res. 2022, 130, 1014–1029. [Google Scholar] [CrossRef]
- Peng, Y.; Wang, W.; Fang, Y.; Hu, H.; Chang, N.; Pang, M.; Hu, Y.F.; Li, X.; Long, H.; Xiong, J.W.; et al. Corrigendum: Inhibition of TGF-β/Smad3 Signaling Disrupts Cardiomyocyte Cell Cycle Progression and Epithelial–Mesenchymal Transition-Like Response During Ventricle Regeneration. Front. Cell Dev. Biol. 2021, 9, 699796. [Google Scholar] [CrossRef]
- Mukherjee, D.; Wagh, G.; Mokalled, M.H.; Kontarakis, Z.; Dickson, A.L.; Rayrikar, A.; Günther, S.; Poss, K.D.; Stainier, D.Y.R.; Patra, C. Ccn2a/Ctgfa is an injury-induced matricellular factor that promotes cardiac regeneration in zebrafish. Development 2020, 148, dev193219. [Google Scholar] [CrossRef]
- Liu, P.; Zhong, T.P. MAPK/ERK signalling is required for zebrafish cardiac regeneration. Biotechnol. Lett. 2017, 39, 1069–1077. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.R.; Wei, T.W.; Tang, C.P.; Sun, J.T.; Shan, T.K.; Fan, Y.; Yang, T.T.; Li, Y.F.; Ma, Y.; Wang, S.B.; et al. MNK2-eIF4E axis promotes cardiac repair in the infarcted mouse heart by activating cyclin D1. J. Mol. Cell. Cardiol. 2022, 166, 91–106. [Google Scholar] [CrossRef] [PubMed]
- Fang, Y.; Lai, K.S.; She, P.; Sun, J.; Tao, W.; Zhong, T.P. Tbx20 Induction Promotes Zebrafish Heart Regeneration by Inducing Cardiomyocyte Dedifferentiation and Endocardial Expansion. Front. Cell Dev. Biol. 2020, 8, 738. [Google Scholar] [CrossRef] [PubMed]
- Koth, J.; Wang, X.; Killen, A.C.; Stockdale, W.T.; Potts, H.G.; Jefferson, A.; Bonkhofer, F.; Riley, P.R.; Patient, R.K.; Göttgens, B.; et al. Runx1 promotes scar deposition and inhibits myocardial proliferation and survival during zebrafish heart regeneration. Development 2020, 147, dev186569. [Google Scholar] [CrossRef] [PubMed]
- Zuppo, D.A.; Missinato, M.A.; Santana-Santos, L.; Li, G.; Benos, P.V.; Tsang, M. Foxm1 regulates cardiomyocyte proliferation in adult zebrafish after cardiac injury. Development 2023, 150, dev201163. [Google Scholar] [CrossRef]
- Wang, Y.; Li, Y.; Feng, J.; Liu, W.; Li, Y.; Liu, J.; Yin, Q.; Lian, H.; Liu, L.; Nie, Y. Mydgf promotes Cardiomyocyte proliferation and Neonatal Heart regeneration. Theranostics 2020, 10, 9100–9112. [Google Scholar] [CrossRef]
- Wang, T.; Zhou, L.Y.; Li, X.M.; Liu, F.; Liang, L.; Chen, X.Z.; Ju, J.; Ponnusamy, M.; Wang, K.; Liu, C.Y.; et al. ABRO1 arrests cardiomyocyte proliferation and myocardial repair by suppressing PSPH. Mol. Ther. 2023, 31, 847–865. [Google Scholar] [CrossRef]
- Gong, R.; Wang, X.; Li, H.; Liu, S.; Jiang, Z.; Zhao, Y.; Yu, Y.; Han, Z.; Yu, Y.; Dong, C.; et al. Loss of m6A methyltransferase METTL3 promotes heart regeneration and repair after myocardial injury. Pharmacol. Res. 2021, 174, 105845. [Google Scholar] [CrossRef]
- An, S.; Chen, Y.; Gao, C.; Qin, B.; Du, X.; Meng, F.; Qi, Y. Inactivation of INK4a and ARF induces myocardial proliferation and improves cardiac repair following ischemia-reperfusion. Mol. Med. Rep. 2015, 12, 5911–5916. [Google Scholar] [CrossRef]
- Beisaw, A.; Kuenne, C.; Guenther, S.; Dallmann, J.; Wu, C.C.; Bentsen, M.; Looso, M.; Stainier, D.Y.R. AP-1 Contributes to Chromatin Accessibility to Promote Sarcomere Disassembly and Cardiomyocyte Protrusion During Zebrafish Heart Regeneration. Circ. Res. 2020, 126, 1760–1778. [Google Scholar] [CrossRef]
- Bühler, A.; Gahr, B.M.; Park, D.D.; Bertozzi, A.; Boos, A.; Dalvoy, M.; Pott, A.; Oswald, F.; Kovall, R.A.; Kühn, B.; et al. Histone deacetylase 1 controls cardiomyocyte proliferation during embryonic heart development and cardiac regeneration in zebrafish. PLoS Genet. 2021, 17, e1009890. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Li, M.; Li, X.; Li, H.; Lai, Y.; Huang, S.; He, X.; Si, X.; Zheng, H.; Liao, W.; et al. Sirt1-inducible deacetylation of p21 promotes cardiomyocyte proliferation. Aging 2019, 11, 12546–12567. [Google Scholar] [CrossRef] [PubMed]
- Ye, S.; Zhao, T.; Zhang, W.; Tang, Z.; Gao, C.; Ma, Z.; Xiong, J.W.; Peng, J.; Tan, W.Q.; Chen, J. p53 isoform Δ113p53 promotes zebrafish heart regeneration by maintaining redox homeostasis. Cell Death Dis. 2020, 11, 568. [Google Scholar] [CrossRef] [PubMed]
- Paddock, S.J.; Swift, S.K.; Alencar-Almeida, V.; Kenarsary, A.; Alvarez-Argote, S.; Flinn, M.A.; Patterson, M.; O’Meara, C.C. IL4Rα signaling promotes neonatal cardiac regeneration and cardiomyocyte cell cycle activity. J. Mol. Cell. Cardiol. 2021, 161, 62–74. [Google Scholar] [CrossRef] [PubMed]
- Zogbi, C.; Oliveira, N.C.; Levy, D.; Bydlowski, S.P.; Bassaneze, V.; Neri, E.A.; Krieger, J.E. Beneficial effects of IL-4 and IL-6 on rat neonatal target cardiac cells. Sci. Rep. 2020, 10, 12350. [Google Scholar] [CrossRef] [PubMed]
- Tang, P.; Ma, S.; Dong, M.; Wang, J.; Chai, S.; Liu, T.; Li, J. Effect of interleukin-6 on myocardial regeneration in mice after cardiac injury. Biomed. Pharmacother. 2018, 106, 303–308. [Google Scholar] [CrossRef]
- Zhao, Y.T.; Wang, J.; Yano, N.; Zhang, L.X.; Wang, H.; Qin, G.; Dubielecka, P.M.; Zhuang, S.; Liu, P.Y.; Eugene, Y.; et al. Irisin promotes cardiac progenitor cell-induced myocardial repair and functional improvement in infarcted heart. J. Cell Physiol. 2019, 234, 1671–1681. [Google Scholar] [CrossRef]
- Frangogiannis, N.G. Cardiac fibrosis: Cell biological mechanisms, molecular pathways and therapeutic opportunities. Mol. Asp. Med. 2019, 65, 70–99. [Google Scholar] [CrossRef]
- Ieda, M.; Tsuchihashi, T.; Ivey, K.N.; Ross, R.S.; Hong, T.T.; Shaw, R.M.; Srivastava, D. Cardiac Fibroblasts Regulate Myocardial Proliferation through β1 Integrin Signaling. Dev. Cell 2009, 16, 233–244. [Google Scholar] [CrossRef]
- Sadahiro, T. Direct Cardiac Reprogramming—Converting Cardiac Fibroblasts to Cardiomyocytes—. Circ. Rep. 2019, 1, 564–567. [Google Scholar] [CrossRef]
- Sadahiro, T.; Yamanaka, S.; Ieda, M. Direct cardiac reprogramming: Progress and challenges in basic biology and clinical applications. Circ. Res. 2015, 116, 1378–1391. [Google Scholar] [CrossRef] [PubMed]
- Di Lullo, G.A.; Sweeney, S.M.; Körkkö, J.; Ala-Kokko, L.; San Antonio, J.D. Mapping the ligand-binding sites and disease-associated mutations on the most abundant protein in the human, type I collagen. J. Biol. Chem. 2002, 277, 4223–4231. [Google Scholar] [CrossRef] [PubMed]
- Sadahiro, T.; Ieda, M. In vivo reprogramming as a new approach to cardiac regenerative therapy. Semin. Cell Dev. Biol. 2022, 122, 21–27. [Google Scholar] [CrossRef] [PubMed]
- Kong, P.; Christia, P.; Frangogiannis, N.G. The pathogenesis of cardiac fibrosis. Cell. Mol. Life Sci. 2014, 71, 549–574. [Google Scholar] [CrossRef]
- Li, L.; Zhao, Q.; Kong, W. Extracellular matrix remodeling and cardiac fibrosis. Matrix Biol. 2018, 68–69, 490–506. [Google Scholar] [CrossRef]
- Cowling, R.T.; Kupsky, D.; Kahn, A.M.; Daniels, L.B.; Greenberg, B.H. Mechanisms of cardiac collagen deposition in experimental models and human disease. Transl. Res. 2019, 209, 138–155. [Google Scholar] [CrossRef]
- Frangogiannis, N.G. The extracellular matrix in myocardial injury, repair, and remodeling. J. Clin. Investig. 2017, 127, 1600–1612. [Google Scholar] [CrossRef]
- Chablais, F.; Veit, J.; Rainer, G.; Jaźwińska, A. The zebrafish heart regenerates after cryoinjury-induced myocardial infarction. BMC Dev. Biol. 2011, 11, 21. [Google Scholar] [CrossRef]
- González, A.; Schelbert, E.B.; Díez, J.; Butler, J. Myocardial Interstitial Fibrosis in Heart Failure: Biological and Translational Perspectives. J. Am. Coll. Cardiol. 2018, 71, 1696–1706. [Google Scholar] [CrossRef]
- Jun, J.I.; Lau, L.F. Resolution of organ fibrosis. J. Clin. Investig. 2018, 128, 97–107. [Google Scholar] [CrossRef]
- Wu, C.C.; Kruse, F.; Vasudevarao, M.D.; Junker, J.P.; Zebrowski, D.C.; Fischer, K.; Noël, E.S.; Grün, D.; Berezikov, E.; Engel, F.B.; et al. Spatially Resolved Genome-wide Transcriptional Profiling Identifies BMP Signaling as Essential Regulator of Zebrafish Cardiomyocyte Regeneration. Dev. Cell 2016, 36, 36–49. [Google Scholar] [CrossRef] [PubMed]
- De Bakker, D.E.M.; Bouwman, M.; Dronkers, E.; Simões, F.C.; Riley, P.R.; Goumans, M.J.; Smits, A.M.; Bakkers, J. Prrx1b restricts fibrosis and promotes Nrg1-dependent cardiomyocyte proliferation during zebrafish heart regeneration. Development 2021, 148, dev198937. [Google Scholar] [CrossRef]
- Notari, M.; Ventura-Rubio, A.; Bedford-Guaus, S.J.; Jorba, I.; Mulero, L.; Navajas, D.; Martí, M.; Raya, A. The local microenvironment limits the regenerative potential of the mouse neonatal heart. Cell Biol. 2018, 4, eaao5553. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Paulsen, M.J.; Hironaka, C.E.; Shin, H.S.; Farry, J.M.; Thakore, A.D.; Jung, J.; Lucian, H.J.; Eskandari, A.; Anilkumar, S.; et al. Natural Heart Regeneration in a Neonatal Rat Myocardial Infarction Model. Cells 2020, 9, 229. [Google Scholar] [CrossRef] [PubMed]
- Nakano, H.; Minami, I.; Braas, D.; Pappoe, H.; Wu, X.; Sagadevan, A.; Vergnes, L.; Fu, K.; Morselli, M.; Dunham, C.; et al. Glucose inhibits cardiac muscle maturation through nucleotide biosynthesis. eLife 2017, 6, e29330. [Google Scholar] [CrossRef]
- Fajardo, V.M.; Feng, I.; Chen, B.Y.; Perez-Ramirez, C.A.; Shi, B.; Clark, P.; Tian, R.; Lien, C.L.; Pellegrini, M.; Christofk, H.; et al. GLUT1 overexpression enhances glucose metabolism and promotes neonatal heart regeneration. Sci. Rep. 2021, 11, 8669. [Google Scholar] [CrossRef]
- Andersen, D.C.; Jensen, C.H.; Baun, C.; Hvidsten, S.; Zebrowski, D.C.; Engel, F.B.; Sheikh, S.P. Persistent scarring and dilated cardiomyopathy suggest incomplete regeneration of the apex resected neonatal mouse myocardium—A 180days follow up study. J. Mol. Cell. Cardiol. 2016, 90, 47–52. [Google Scholar] [CrossRef]
- Sarohi, V.; Srivastava, S.; Basak, T. Comprehensive Mapping and Dynamics of Site-Specific Prolyl-Hydroxylation, Lysyl-Hydroxylation and Lysyl O-Glycosylation of Collagens Deposited in ECM During Zebrafish Heart Regeneration. Front. Mol. Biosci. 2022, 9, 892763. [Google Scholar] [CrossRef]
- Akhurst, R.J.; Hata, A. Targeting the TGFβ signalling pathway in disease. Nat. Rev. Drug Discov. 2012, 11, 790–811. [Google Scholar] [CrossRef]
- Massagué, J. TGFβ signalling in context. Nat. Rev. Mol. Cell Biol. 2012, 13, 616–630. [Google Scholar] [CrossRef]
- Lal, H.; Ahmad, F.; Zhou, J.; Yu, J.E.; Vagnozzi, R.J.; Guo, Y.; Yu, D.; Tsai, E.J.; Woodgett, J.; Gao, E.; et al. Cardiac fibroblast glycogen synthase kinase-3β regulates ventricular remodeling and dysfunction in ischemic heart. Circulation 2014, 130, 419–430. [Google Scholar] [CrossRef]
- Zhou, Q.; Meng, D.; Li, F.; Zhang, X.; Liu, L.; Zhu, Y.; Liu, S.; Xu, M.; Deng, J.; Lei, Z.; et al. Inhibition of HIPK2 protects stress-induced pathological cardiac remodeling. Ebiomedicine 2022, 85, 104274. [Google Scholar] [CrossRef]
- Ni, C.; Chen, Y.; Xu, Y.; Zhao, J.; Li, Q.; Xiao, C.; Wu, Y.; Wang, J.; Wang, Y.; Zhong, Z.; et al. FMO2 (Flavin Containing Monooxygenase 2) Prevents Cardiac Fibrosis via CYP2J3-SMURF2 Axis. Circ. Res. 2022, 131, 189–206. [Google Scholar] [CrossRef] [PubMed]
- Li, C.Y.; Zhang, J.R.; Li, X.X.; Zhao, L.; Xi, H.; Hu, W.N.; Li, S.N. Lefty1 Ameliorates Post-infarction Fibrosis by Suppressing p-Smad2 and p-ERK1/2 Signaling Pathways. J. Cardiovasc. Transl. Res. 2021, 14, 636–646. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.; Lei, H.; Wang, J.Y.; Zhang, C.L.; Feng, H.; Fu, F.Y.; Li, L.; Wu, L.L. CTRP3 attenuates post-infarct cardiac fibrosis by targeting Smad3 activation and inhibiting myofibroblast differentiation. J. Mol. Med. 2015, 93, 1311–1325. [Google Scholar] [CrossRef] [PubMed]
- Jin, D.; Han, F. FOXF1 ameliorates angiotensin II-induced cardiac fibrosis in cardiac fibroblasts through inhibiting the TGF-β1/Smad3 signaling pathway. J. Recept. Signal Transduct. 2020, 40, 493–500. [Google Scholar] [CrossRef]
- Lu, Y.G.; Tan, H.; Ma, Q.; Li, X.X.; Cui, J.; Zhang, X.; Liang, X.L.; Tie, Y.Q. SH2 domain-containing protein tyrosine phosphatase-2 (SHP-2) prevents cardiac remodeling after myocardial infarction through ERK/SMAD signaling pathway. Human. Cell 2021, 34, 325–334. [Google Scholar] [CrossRef]
- Humeres, C.; Shinde, A.V.; Hanna, A.; Alex, L.; Hernández, S.C.; Li, R.; Chen, B.; Conway, S.J.; Frangogiannis, N.G. Smad7 effects on TGF-β and ErbB2 restrain myofibroblast activation and protect from postinfarction heart failure. J. Clin. Investig. 2022, 132, e146926. [Google Scholar] [CrossRef]
- Wu, Y.; Chen, S.; Wen, P.; Wu, M.; Wu, Y.; Mai, M.; Huang, J. PGAM1 deficiency ameliorates myocardial infarction remodeling by targeting TGF-β via the suppression of inflammation, apoptosis and fibrosis. Biochem. Biophys. Res. Commun. 2021, 534, 933–940. [Google Scholar] [CrossRef]
- Wang, B.; Tan, Y.; Zhou, W.; Yang, J.; Jiang, Y.; Liu, X.; Zhan, Z. Loss of BTK ameliorates the pathological cardiac fibrosis and dysfunction. Matrix Biol. 2022, 112, 171–189. [Google Scholar] [CrossRef]
- Li, F.; Li, L.; Zhang, J.; Yang, X.; Liu, Y. Histone methyltransferase DOT1L mediates the TGF-β1/Smad3 signaling pathway through epigenetic modification of SYK in myocardial infarction. Human. Cell 2022, 35, 98–110. [Google Scholar] [CrossRef] [PubMed]
- Jurado Acosta, A.; Rysä, J.; Szabo, Z.; Moilanen, A.M.; Komati, H.; Nemer, M.; Ruskoaho, H. Transcription factor PEX1 modulates extracellular matrix turnover through regulation of MMP-9 expression. Cell Tissue Res. 2017, 367, 369–385. [Google Scholar] [CrossRef] [PubMed]
- Wu, A.; Zhang, L.; Chen, J.; Li, H.; Yang, P.; Chen, M.; Liu, Q. Limb-bud and Heart (LBH) mediates proliferation, fibroblast-to-myofibroblast transition and EMT-like processes in cardiac fibroblasts. Mol. Cell. Biochem. 2021, 476, 2685–2701. [Google Scholar] [CrossRef] [PubMed]
- Gao, A.E.; Sullivan, K.E.; Black, L.D. Lysyl oxidase expression in cardiac fibroblasts is regulated by α2β1 integrin interactions with the cellular microenvironment. Biochem. Biophys. Res. Commun. 2016, 475, 70–75. [Google Scholar] [CrossRef]
- Tian, X.; Sun, C.; Wang, X.; Ma, K.; Chang, Y.; Guo, Z.; Si, J. ANO1 regulates cardiac fibrosis via ATI-mediated MAPK pathway. Cell Calcium. 2020, 92, 102306. [Google Scholar] [CrossRef]
- Zhang, Q.J.; He, Y.; Li, Y.; Shen, H.; Lin, L.; Zhu, M.; Wang, Z.; Luo, X.; Hill, J.A.; Cao, D.; et al. Matricellular Protein Cilp1 Promotes Myocardial Fibrosis in Response to Myocardial Infarction. Circ. Res. 2021, 129, 1021–1035. [Google Scholar] [CrossRef]
- Lei, H.; Wu, D.; Wang, J.Y.; Li, L.; Zhang, C.L.; Feng, H.; Fu, F.Y.; Wu, L.L. C1q/tumor necrosis factor-related protein-6 attenuates post-infarct cardiac fibrosis by targeting RhoA/MRTF-A pathway and inhibiting myofibroblast differentiation. Basic Res. Cardiol. 2015, 110, 35. [Google Scholar] [CrossRef]
- Scharf, G.M.; Kilian, K.; Cordero, J.; Wang, Y.; Grund, A.; Hofmann, M.; Froese, N.; Wang, X.; Kispert, A.; Kist, R.; et al. Inactivation of Sox9 in fibroblasts reduces cardiac fibrosis and inflammation. JCI Insight 2019, 4, 126721. [Google Scholar] [CrossRef]
- Li, M.; Wu, J.; Hu, G.; Song, Y.; Shen, J.; Xin, J.; Li, Z.; Liu, W.; Dong, E.; Xu, M.; et al. Pathological matrix stiffness promotes cardiac fibroblast differentiation through the POU2F1 signaling pathway. Sci. China Life Sci. 2021, 64, 242–254. [Google Scholar] [CrossRef]
- Song, K.; Nam, Y.J.; Luo, X.; Qi, X.; Tan, W.; Huang, G.N.; Acharya, A.; Smith, C.L.; Tallquist, M.D.; Neilson, E.G.; et al. Heart repair by reprogramming non-myocytes with cardiac transcription factors. Nature 2012, 485, 599–604. [Google Scholar] [CrossRef]
- Santiago, J.J.; McNaughton, L.J.; Koleini, N.; Ma, X.; Bestvater, B.; Nickel, B.E.; Fandrich, R.R.; Wigle, J.T.; Freed, D.H.; Arora, R.C.; et al. High molecular weight fibroblast growth factor-2 in the human heart is a potential target for prevention of cardiac remodeling. PLoS ONE 2014, 9, e97281. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Yin, H.; Jiao, L.; Liu, T.; Gao, Y.; Shao, Y.; Zhang, Y.; Shan, H.; Zhang, Y.; Yang, B. Abnormal Downregulation of Caveolin-3 Mediates the Pro-Fibrotic Action of MicroRNA-22 in a Model of Myocardial Infarction. Cell. Physiol. Biochem. 2018, 45, 1641–1653. [Google Scholar] [CrossRef] [PubMed]
- Dufeys, C.; Daskalopoulos, E.P.; Castanares-Zapatero, D.; Conway, S.J.; Ginion, A.; Bouzin, C.; Ambroise, J.; Bearzatto, B.; Gala, J.L.; Heymans, S.; et al. AMPKα1 deletion in myofibroblasts exacerbates post-myocardial infarction fibrosis by a connexin 43 mechanism. Basic Res. Cardiol. 2021, 116, 10. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Villalba, A.; Romero, J.P.; Hernández, S.C.; Vilas-Zornoza, A.; Fortelny, N.; Castro-Labrador, L.; San Martin-Uriz, P.; Lorenzo-Vivas, E.; García-Olloqui, P.; Palacio, M.; et al. Single-Cell RNA Sequencing Analysis Reveals a Crucial Role for CTHRC1 (Collagen Triple Helix Repeat Containing 1) Cardiac Fibroblasts After Myocardial Infarction. Circulation 2020, 142, 1831–1847. [Google Scholar] [CrossRef] [PubMed]
- Francisco, J.; Zhang, Y.; Jeong, J.I.; Mizushima, W.; Ikeda, S.; Ivessa, A.; Oka, S.; Zhai, P.; Tallquist, M.D.; Del Re, D.P. Blockade of Fibroblast YAP Attenuates Cardiac Fibrosis and Dysfunction Through MRTF-A Inhibition. JACC Basic Transl. Sci. 2020, 5, 931–945. [Google Scholar] [CrossRef]
- Sun, Y.; Ma, M.; Cao, D.; Zheng, A.; Zhang, Y.; Su, Y.; Wang, J.; Xu, Y.; Zhou, M.; Tang, Y.; et al. Inhibition of Fap Promotes Cardiac Repair by Stabilizing BNP. Circ. Res. 2023, 132, 586–600. [Google Scholar] [CrossRef]
- Horii, Y.; Matsuda, S.; Toyota, C.; Morinaga, T.; Nakaya, T.; Tsuchiya, S.; Ohmuraya, M.; Hironaka, T.; Yoshiki, R.; Kasai, K.; et al. VGLL3 is a mechanosensitive protein that promotes cardiac fibrosis through liquid–liquid phase separation. Nat. Commun. 2023, 14, 550. [Google Scholar] [CrossRef]
- Li, T.; Zhuang, Y.; Yang, W.; Xie, Y.; Shang, W.; Su, S.; Dong, X.; Wu, J.; Jiang, W.; Zhou, Y.; et al. Silencing of METTL3 attenuates cardiac fibrosis induced by myocardial infarction via inhibiting the activation of cardiac fibroblasts. FASEB J. 2021, 35, e21162. [Google Scholar] [CrossRef]
- Zhuang, Y.; Li, T.; Hu, X.; Xie, Y.; Pei, X.; Wang, C.; Li, Y.; Liu, J.; Tian, Z.; Zhang, X.; et al. MetBil as a novel molecular regulator in ischemia-induced cardiac fibrosis via METTL3-mediated m6A modification. FASEB J. 2023, 37, e22797. [Google Scholar] [CrossRef]
- Yao, H.; He, Q.; Huang, C.; Wei, S.; Gong, Y.; Li, X.; Liu, W.; Xu, Z.; Wu, H.; Zheng, C.; et al. Panaxatriol saponin ameliorates myocardial infarction-induced cardiac fibrosis by targeting Keap1/Nrf2 to regulate oxidative stress and inhibit cardiac-fibroblast activation and proliferation. Free. Radic. Biol. Med. 2022, 190, 264–275. [Google Scholar] [CrossRef]
- Zhang, M.; Zhou, T.; Liu, M.; Xia, N.; Gu, M.; Tang, T.; Nie, S.; Zhu, Z.; Lv, B.; Jiao, J.; et al. Inhibition of fibroblast IL-6 production by ACKR4 deletion alleviates cardiac remodeling after myocardial infarction. Biochem. Biophys. Res. Commun. 2021, 547, 139–147. [Google Scholar] [CrossRef]
- Song, J.; Frieler, R.A.; Whitesall, S.E.; Chung, Y.; Vigil, T.M.; Muir, L.A.; Ma, J.; Brombacher, F.; Goonewardena, S.N.; Lumeng, C.N.; et al. Myeloid interleukin-4 receptor α is essential in postmyocardial infarction healing by regulating inflammation and fibrotic remodeling. Am. J. Physiol.-Heart Circ. Physiol. 2021, 320, H323–37. [Google Scholar] [CrossRef]
- Wang, J.; Wang, M.; Lu, X.; Zhang, Y.; Zeng, S.; Pan, X.; Zhou, Y.; Wang, H.; Chen, N.; Cai, F.; et al. IL-6 inhibitors effectively reverse post-infarction cardiac injury and ischemic myocardial remodeling via the TGF-β1/Smad3 signaling pathway. Exp. Ther. Med. 2022, 24, 576. [Google Scholar] [CrossRef] [PubMed]
- Mia, M.M.; Cibi, D.M.; Ghani, S.A.B.A.; Song, W.; Tee, N.; Ghosh, S.; Mao, J.; Olson, E.N.; Singh, M.K. YAP/TAZ deficiency reprograms macrophage phenotype and improves infarct healing and cardiac function after myocardial infarction. PLoS Biol. 2020, 18, e3000941. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Chen, Y.; Zhao, Z.; Meng, Q.; Yu, Y.; Sun, J.; Yang, Z.; Chen, Y.; Li, J.; Ma, T.; et al. Engineered exosomes with ischemic myocardium-targeting peptide for targeted therapy in myocardial infarction. J. Am. Heart Assoc. 2018, 7, e008737. [Google Scholar] [CrossRef] [PubMed]
- Vandergriff, A.; Huang, K.; Shen, D.; Hu, S.; Hensley, M.T.; Caranasos, T.G.; Qian, L.; Cheng, K. Targeting regenerative exosomes to myocardial infarction using cardiac homing peptide. Theranostics 2018, 8, 1869–1878. [Google Scholar] [CrossRef]
- Riaud, M.; Hilairet, G.; Sindji, L.; Perdomo, L.; Montero-Menei, C.N.; Martinez, M.C. Pharmacology active microcarriers delivering HGF associated with extracellular vesicles for myocardial repair. Eur. J. Pharm. Biopharm. 2021, 169, 268–279. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.; Fu, W.; Li, L.; Xia, X.; Liao, Q.; Yue, R.; Chen, H.; Chen, X.; An, S.; Zeng, C.; et al. Correction: Therapeutic effect of a novel Wnt pathway inhibitor on cardiac regeneration after myocardial infarction. Clin. Sci. 2017, 131, 22919–22932, Erratum in Clin. Sci. 2019, 133, 149. [Google Scholar] [CrossRef]
- Wang, Y.B.; Ma, S.; Wang, Q.; Hu, W.X.; Wang, D.J.; Li, X.J.; Su, T.; Qin, X.; Zhang, X.T.; Ma, K.; et al. Effects of cannabinoid receptor type 2 on endogenous myocardial regeneration by activating cardiac progenitor cells in mouse infarcted heart. Sci. China Life Sci. 2014, 57, 201–208. [Google Scholar] [CrossRef]
- Zhao, N.; Yu, H.Y.; Yu, H.T.; Sun, M.; Zhang, Y.Y.; Xu, M.; Gao, W. miRNA-711-SP1-collagen-I pathway is involved in the anti-fibrotic effect of pioglitazone in myocardial infarction. Sci. China Life Sci. 2013, 56, 431–439. [Google Scholar] [CrossRef]
- Song, B.W.; Kim, S.; Kim, R.; Jeong, S.; Moon, H.; Kim, H.; Vasileva, E.A.; Mishchenko, N.P.; Fedoreyev, S.A.; Stonik, V.A.; et al. Regulation of Inflammation-Mediated Endothelial to Mesenchymal Transition with Echinochrome a for Improving Myocardial Dysfunction. Mar. Drugs 2022, 20, 756. [Google Scholar] [CrossRef] [PubMed]
- Du, J.; Zheng, L.; Gao, P.; Yang, H.; Yang, W.J.; Guo, F.; Liang, R.; Feng, M.; Wang, Z.; Zhang, Z.; et al. A small-molecule cocktail promotes mammalian cardiomyocyte proliferation and heart regeneration. Cell Stem Cell 2022, 29, 545–558.e13. [Google Scholar] [CrossRef]
- Tripathi, H.; Domingues, A.; Donahue, R.; Cras, A.; Guerin, C.L.; Gao, E.; Levitan, B.; Ratajczak, M.Z.; Smadja, D.M.; Abdel-Latif, A.; et al. Combined Transplantation of Human MSCs and ECFCs Improves Cardiac Function and Decrease Cardiomyocyte Apoptosis After Acute Myocardial Infarction. Stem Cell Rev. Rep. 2023, 19, 573–577. [Google Scholar] [CrossRef] [PubMed]
- Li, S.H.; Sun, Z.; Guo, L.; Han, M.; Wood, M.F.G.; Ghosh, N.; Alex Vitkin, I.; Weisel, R.D.; Li, R.K. Elastin overexpression by cell-based gene therapy preserves matrix and prevents cardiac dilation. J. Cell. Mol. Med. 2012, 16, 2429–2439. [Google Scholar] [CrossRef]
- Van Laake, L.W.; Van Donselaar, E.G.; Monshouwer-Kloots, J.; Schreurs, C.; Passier, R.; Humbel, B.M.; Doevendans, P.A.; Sonnenberg, A.; Verkleij, A.J.; Mummery, C.L. Extracellular matrix formation after transplantation of human embryonic stem cell-derived cardiomyocytes. Cell Mol. Life Sci. 2010, 67, 277–290. [Google Scholar] [CrossRef]
- Li, P.; Hu, J.; Wang, J.; Zhang, J.; Wang, L.; Zhang, C. The Role of Hydrogel in Cardiac Repair and Regeneration for Myocardial Infarction: Recent Advances and Future Perspectives. Bioengineering 2023, 10, 165. [Google Scholar] [CrossRef]
- Sun, X.; Nunes, S.S. Bioengineering Approaches to Mature Human Pluripotent Stem Cell-Derived Cardiomyocytes. Front. Cell Dev. Biol. 2017, 5, 19. [Google Scholar] [CrossRef]
- Chen, X.; Zhu, L.; Wang, X.; Xiao, J. Insight into Heart-Tailored Architectures of Hydrogel to Restore Cardiac Functions after Myocardial Infarction. Mol. Pharm. 2023, 20, 57–81. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhang, J.; Qin, Z.; Fan, Z.; Lu, C.; Chen, B.; Zhao, J.; Li, X.; Xiao, F.; Lin, X.; et al. Preparation of high bioactivity multilayered bone-marrow mesenchymal stem cell sheets for myocardial infarction using a 3D-dynamic system. Acta Biomater. 2018, 72, 182–195. [Google Scholar] [CrossRef]
- Guan, J.; Wang, F.; Li, Z.; Chen, J.; Guo, X.; Liao, J.; Moldovan, N.I. The stimulation of the cardiac differentiation of mesenchymal stem cells in tissue constructs that mimic myocardium structure and biomechanics. Biomaterials 2011, 32, 5568–5580. [Google Scholar] [CrossRef]
- Amin, S.; Banijamali, S.E.; Tafazoli-Shadpour, M.; Shokrgozar, M.A.; Dehghan, M.M.; Haghighipour, N.; Mahdian, R.; Bayati, V.; Abbasnia, P. Comparing the effect of equiaxial cyclic mechanical stimulation on GATA4 expression in adipose-derived and bone marrow-derived mesenchymal stem cells. Cell Biol. Int. 2014, 38, 219–227. [Google Scholar] [CrossRef] [PubMed]
- Van Marion, M.H.; Bax, N.A.M.; Van Turnhout, M.a.r.k.C.; Mauretti, A.; Van Der Schaft, D.W.J.; Goumans, M.J.o.s.é.T.H.; Bouten, C.V.C. Behavior of CMPCs in unidirectional constrained and stress-free 3D hydrogels. J. Mol. Cell. Cardiol. 2015, 87, 79–91. [Google Scholar] [CrossRef] [PubMed]
- Mauretti, A.; Bax, N.A.M.; Van Marion, M.H.; Goumans, M.J.; Sahlgren, C.; Bouten, C.V.C. Cardiomyocyte progenitor cell mechanoresponse unrevealed: Strain avoidance and mechanosome development. Integr. Biol. 2016, 8, 991–1001. [Google Scholar] [CrossRef]
- Shams, Z.; Akbari, B.; Aghdami, N. Bioinspired Device Improves The Cardiogenic Potential of Cardiac Progenitor Cells. Cell J. 2021, 23, 129–136. [Google Scholar] [CrossRef]
- Chimenti, I.; Rizzitelli, G.; Gaetani, R.; Angelini, F.; Ionta, V.; Forte, E.; Frati, G.; Schussler, O.; Barbetta, A.; Messina, E.; et al. Human cardiosphere-seeded gelatin and collagen scaffolds as cardiogenic engineered bioconstructs. Biomaterials 2011, 32, 9271–9281. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, I.; Carrion, K.; Serrano, R.; Dyo, J.; Sasik, R.; Lund, S.; Willems, E.; Aceves, S.; Meili, R.; Mercola, M.; et al. Cyclic stretch of embryonic cardiomyocytes increases proliferation, growth, and expression while repressing Tgf-β signaling. J. Mol. Cell. Cardiol. 2015, 79, 133–144. [Google Scholar] [CrossRef]
- Shi, T.; Wang, P.; Ren, Y.; Zhang, W.; Ma, J.; Li, S.; Tan, X.; Chi, B. Conductive Hydrogel Patches with High Elasticity and Fatigue Resistance for Cardiac Microenvironment Remodeling. ACS Appl. Mater. Interfaces 2023, 15, 14005–14018. [Google Scholar] [CrossRef]
- Abilez, O.J.; Tzatzalos, E.; Yang, H.; Zhao, M.T.; Jung, G.; Zöllner, A.M.; Tiburcy, M.; Riegler, J.; Matsa, E.; Shukla, P.; et al. Passive Stretch Induces Structural and Functional Maturation of Engineered Heart Muscle as Predicted by Computational Modeling. Stem Cells 2018, 36, 265–277. [Google Scholar] [CrossRef]
- Mihic, A.; Li, J.; Miyagi, Y.; Gagliardi, M.; Li, S.H.; Zu, J.; Weisel, R.D.; Keller, G.; Li, R.K. The effect of cyclic stretch on maturation and 3D tissue formation of human embryonic stem cell-derived cardiomyocytes. Biomaterials 2014, 35, 2798–2808. [Google Scholar] [CrossRef]
- Ahadian, S.; Yamada, S.; Ramón-Azcón, J.; Estili, M.; Liang, X.; Nakajima, K.; Shiku, H.; Khademhosseini, A.; Matsue, T. Hybrid hydrogel-aligned carbon nanotube scaffolds to enhance cardiac differentiation of embryoid bodies. Acta Biomater. 2016, 31, 134–143. [Google Scholar] [CrossRef]
- Ravishankar, P.; Tandon, I.; Balachandran, K. Effect of Cyclic Uniaxial Mechanical Strain on Endothelial Progenitor Cell Differentiation. Cardiovasc. Eng. Technol. 2022, 13, 872–885. [Google Scholar] [CrossRef] [PubMed]
- Brougham, C.M.; Levingstone, T.J.; Jockenhoevel, S.; Flanagan, T.C.; O’Brien, F.J. Incorporation of fibrin into a collagen–glycosaminoglycan matrix results in a scaffold with improved mechanical properties and enhanced capacity to resist cell-mediated contraction. Acta Biomater. 2015, 26, 205–214. [Google Scholar] [CrossRef]
- Cheng, M.; Moretti, M.; Engelmayr, G.C.; Freed, L.E. Insulin-like Growth Factor-I and Slow, Bi-directional Perfusion Enhance the Formation of Tissue-Engineered Cardiac Grafts. Tissue Eng. Part A 2009, 15, 645–653. [Google Scholar] [CrossRef]
- Gao, L.; Gregorich, Z.R.; Zhu, W.; Mattapally, S.; Oduk, Y.; Lou, X.; Kannappan, R.; Borovjagin, A.V.; Walcott, G.P.; Pollard, A.E.; et al. Large Cardiac Muscle Patches Engineered From Human Induced-Pluripotent Stem Cell–Derived Cardiac Cells Improve Recovery From Myocardial Infarction in Swine. Circulation 2018, 137, 1712–1730. [Google Scholar] [CrossRef] [PubMed]
- Shachar, M.; Benishti, N.; Cohen, S. Effects of mechanical stimulation induced by compression and medium perfusion on cardiac tissue engineering. Biotechnol. Prog. 2012, 28, 1551–1559. [Google Scholar] [CrossRef] [PubMed]
- Ahadian, S.; Davenport Huyer, L.; Estili, M.; Yee, B.; Smith, N.; Xu, Z.; Sun, Y.; Radisic, M. Moldable elastomeric polyester-carbon nanotube scaffolds for cardiac tissue engineering. Acta Biomater. 2017, 52, 81–91. [Google Scholar] [CrossRef]
- Pretorius, D.; Kahn-Krell, A.M.; LaBarge, W.C.; Lou, X.; Kannappan, R.; Pollard, A.E.; Fast, V.G.; Berry, J.L.; Eberhardt, A.W.; Zhang, J. Fabrication and characterization of a thick, viable bi-layered stem cell-derived surrogate for future myocardial tissue regeneration. Biomed. Mater. 2021, 16, 035007. [Google Scholar] [CrossRef]
- Lux, M.; Andrée, B.; Horvath, T.; Nosko, A.; Manikowski, D.; Hilfiker-Kleiner, D.; Haverich, A.; Hilfiker, A. In vitro maturation of large-scale cardiac patches based on a perfusable starter matrix by cyclic mechanical stimulation. Acta Biomater. 2016, 30, 177–187. [Google Scholar] [CrossRef]
- Zhou, Q.; Li, L.; Zhao, B.; Guan, K.L. The Hippo Pathway in Heart Development, Regeneration, and Diseases. Circ. Res. 2015, 116, 1431–1447. [Google Scholar] [CrossRef]
- Llucià-Valldeperas, A.; Bragós, R.; Soler-Botija, C.; Roura, S.; Gálvez-Montón, C.; Prat-Vidal, C.; Perea-Gil, I.; Bayes-Genis, A. Unravelling the effects of mechanical physiological conditioning on cardiac adipose tissue-derived progenitor cells in vitro and in silico. Sci. Rep. 2018, 8, 499. [Google Scholar] [CrossRef]
- Cassino, T.R.; Drowley, L.; Okada, M.; Beckman, S.A.; Keller, B.; Tobita, K.; LeDuc, P.R.; Huard, J. Mechanical Loading of Stem Cells for Improvement of Transplantation Outcome in a Model of Acute Myocardial Infarction: The Role of Loading History. Tissue Eng. Part A 2012, 18, 1101–1108. [Google Scholar] [CrossRef] [PubMed]
- Zimmerman, S.D.; Karlon, W.J.; Holmes, J.W.; Omens, J.H.; Covell, J.W. Structural and mechanical factors influencing infarct scar collagen organization. Am. J. Physiol.-Heart Circ. Physiol. 2000, 278, H194–200. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Wisneski, A.; Imbrie-Moore, A.M.; Paulsen, M.J.; Wang, Z.; Xuan, Y.; Lopez Hernandez, H.; Hironaka, C.E.; Lucian, H.J.; Shin, H.S.; et al. Natural cardiac regeneration conserves native biaxial left ventricular biomechanics after myocardial infarction in neonatal rats. J. Mech. Behav. Biomed. Mater. 2022, 126, 105074. [Google Scholar] [CrossRef] [PubMed]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Castillo-Casas, J.M.; Caño-Carrillo, S.; Sánchez-Fernández, C.; Franco, D.; Lozano-Velasco, E. Comparative Analysis of Heart Regeneration: Searching for the Key to Heal the Heart—Part II: Molecular Mechanisms of Cardiac Regeneration. J. Cardiovasc. Dev. Dis. 2023, 10, 357. https://doi.org/10.3390/jcdd10090357
Castillo-Casas JM, Caño-Carrillo S, Sánchez-Fernández C, Franco D, Lozano-Velasco E. Comparative Analysis of Heart Regeneration: Searching for the Key to Heal the Heart—Part II: Molecular Mechanisms of Cardiac Regeneration. Journal of Cardiovascular Development and Disease. 2023; 10(9):357. https://doi.org/10.3390/jcdd10090357
Chicago/Turabian StyleCastillo-Casas, Juan Manuel, Sheila Caño-Carrillo, Cristina Sánchez-Fernández, Diego Franco, and Estefanía Lozano-Velasco. 2023. "Comparative Analysis of Heart Regeneration: Searching for the Key to Heal the Heart—Part II: Molecular Mechanisms of Cardiac Regeneration" Journal of Cardiovascular Development and Disease 10, no. 9: 357. https://doi.org/10.3390/jcdd10090357