
Structural Evaluation and Conformational Dynamics of ZNF141T474I Mutation Provoking Postaxial Polydactyly Type A

 , and
, and
Abstract
:1. Introduction
2. Methodologies
2.1. SNP’s Annotation
2.2. ZNF141WT 3D Structure Prediction
2.3. Protein Model Refinement and Validation
2.4. ZNF141T474I Structure Prediction and Comparison with the ZNF141WT Structure
2.5. Conservation Analysis
2.6. Molecular Dynamics Simulations
3. Results
3.1. SNPs Annotation
3.2. Structure Prediction of C2-H2 Domain (171-474) of ZNF141 Protein
3.3. Validation of 3D Structure
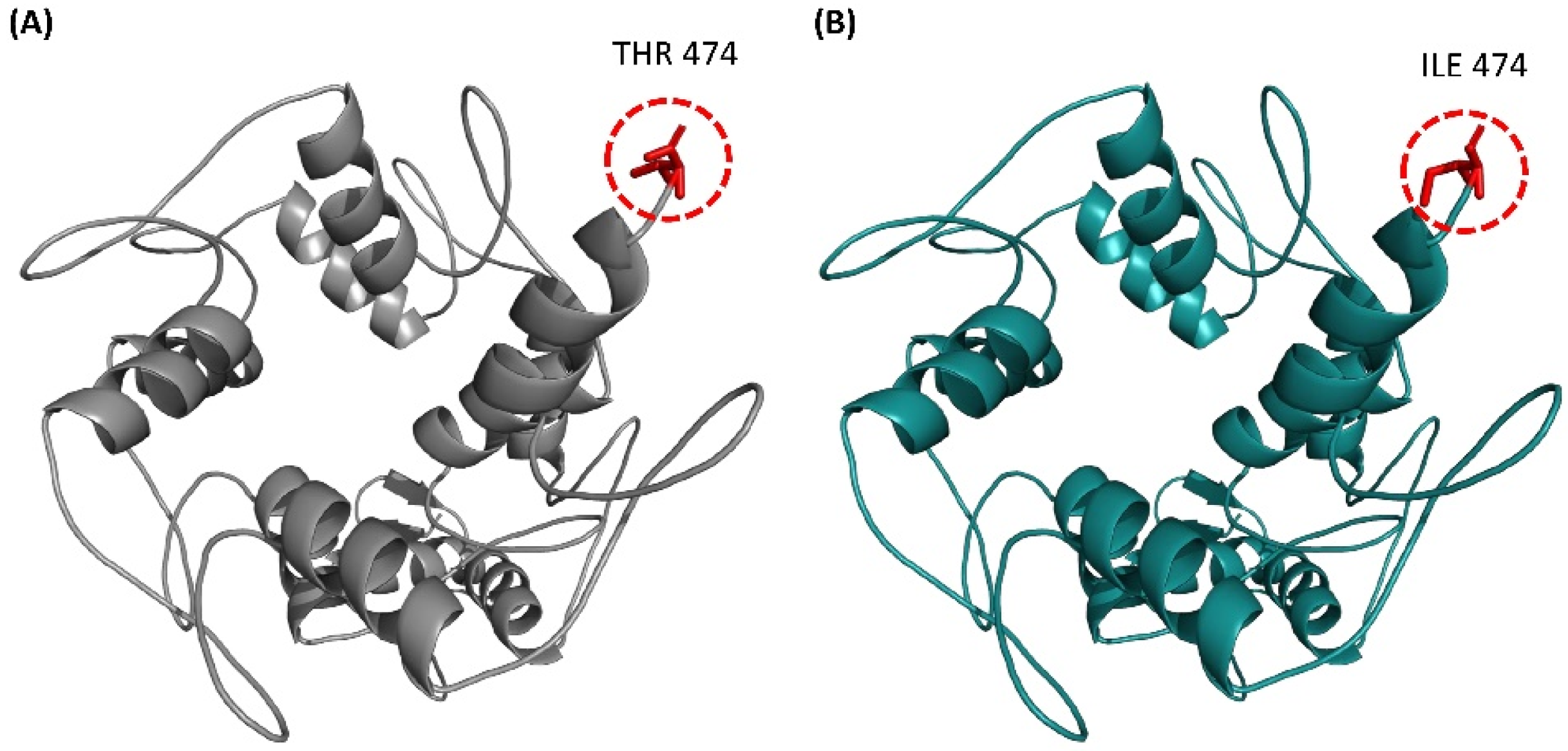
3.4. Structural Comparison of ZNF141WT and ZNF141T474I
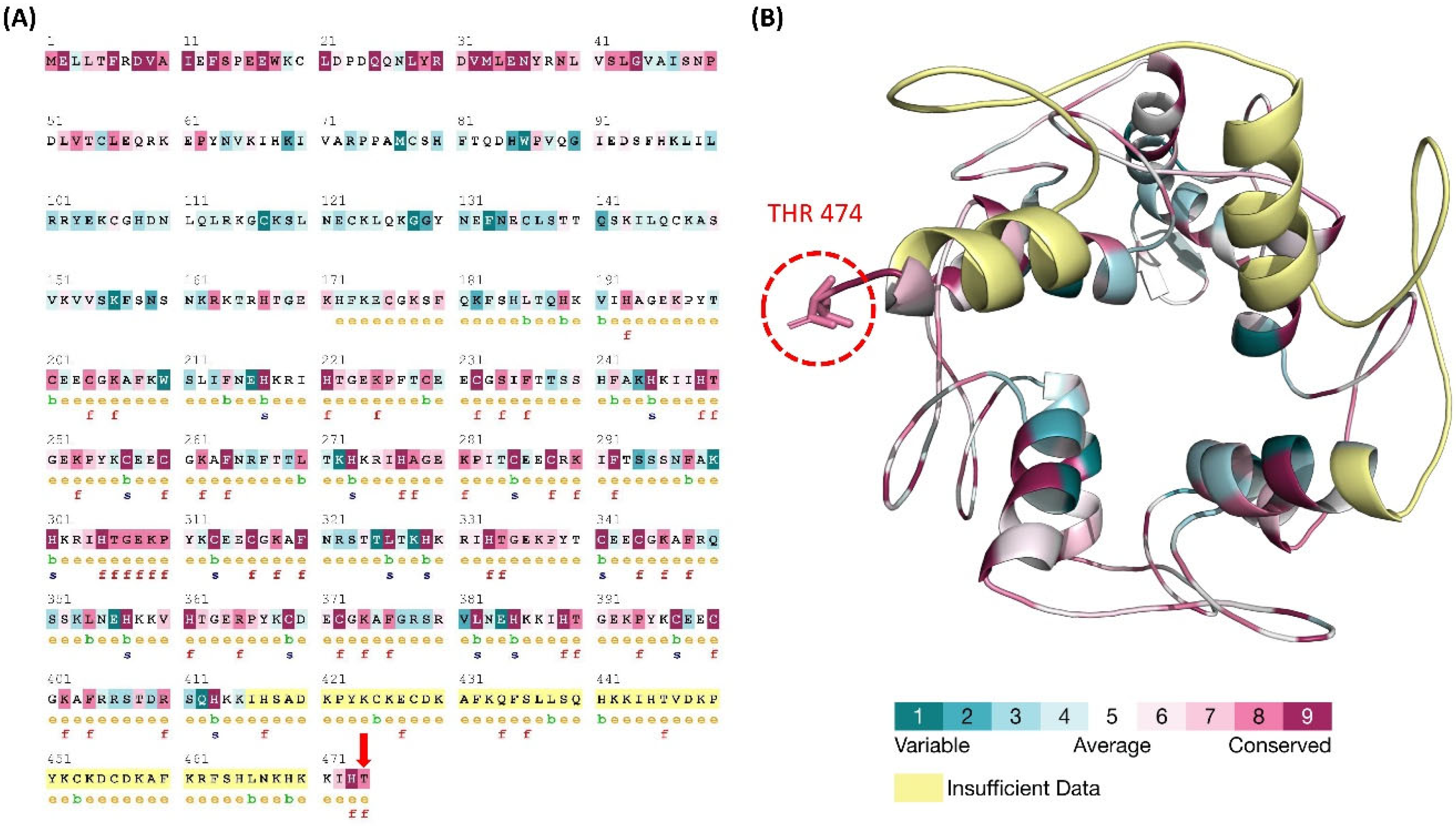
3.5. ConSurf Conservation Analysis
3.6. Molecular Dynamics Simulation Analysis
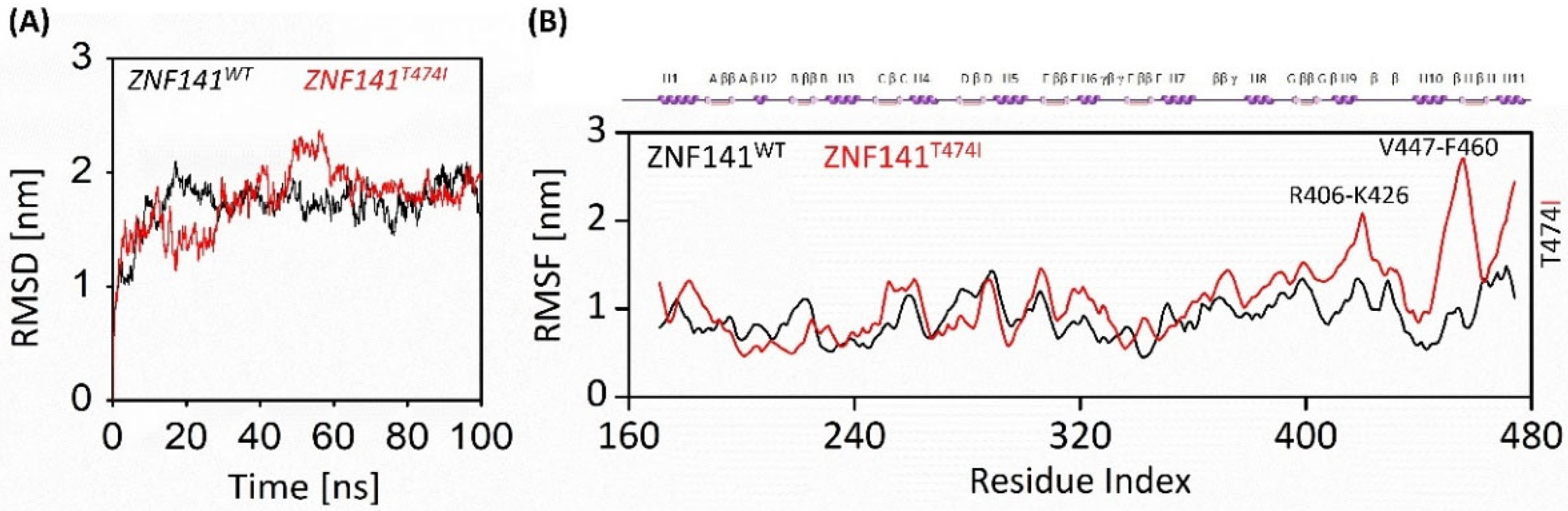
3.6.1. Stability of ZNF141 Protein
3.6.2. Flexibility Analysis of ZNF141 Protein
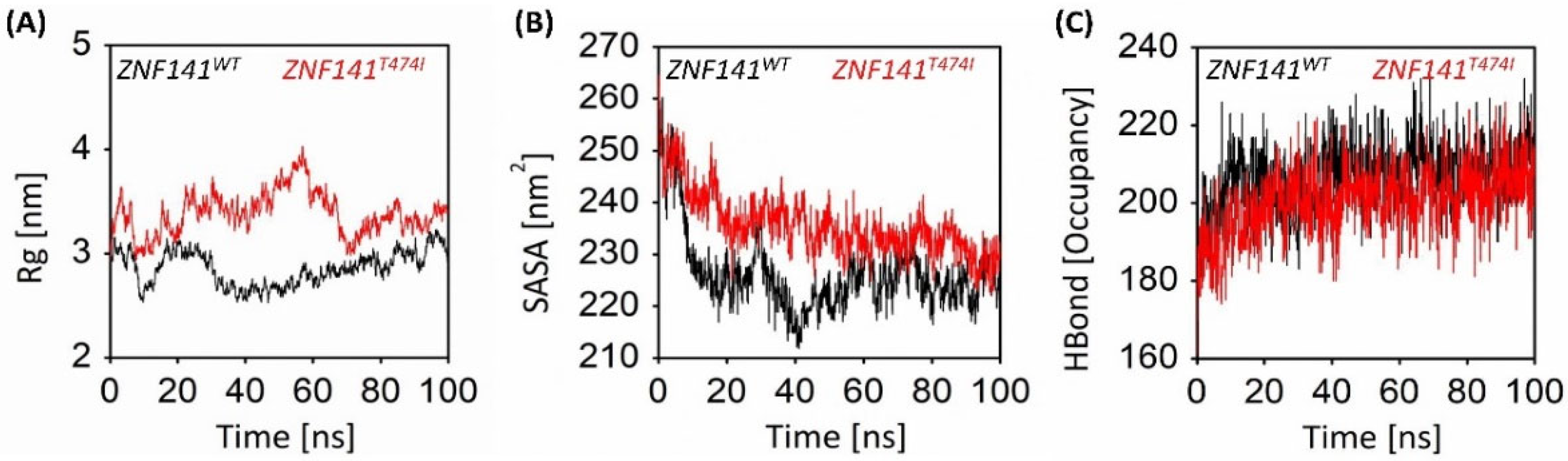
3.6.3. Gyration Analysis of ZNF141 Protein
3.6.4. Solvent Accessible Surface Area Analysis
3.6.5. Intra-Protein Hydrogen Bond Analysis
3.7. Secondary Structure Analysis
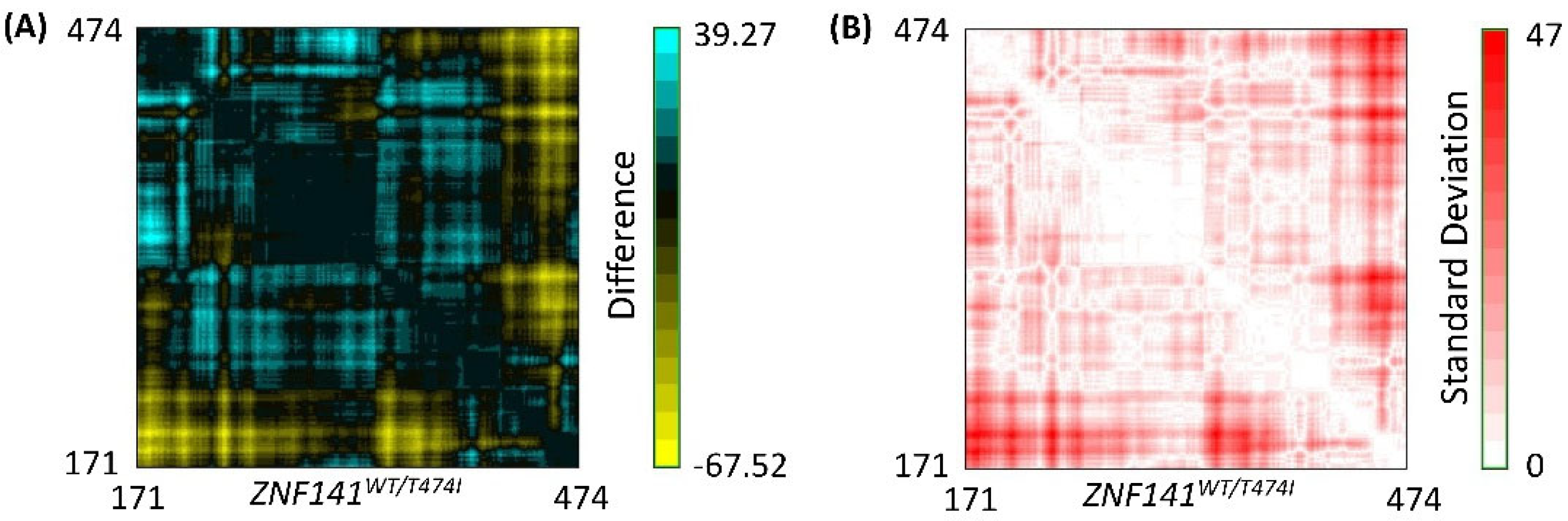
3.8. Residue to Residue Distance Map
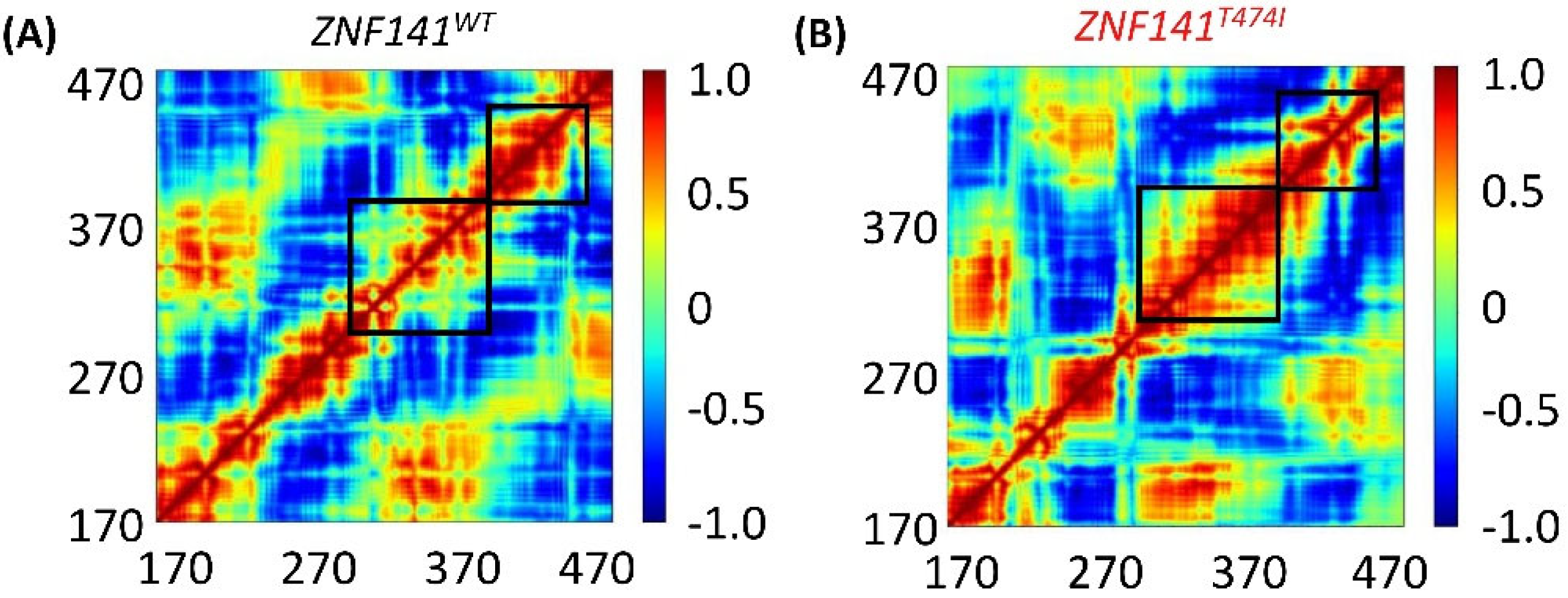
3.9. Dynamic Cross Correlation Map (DCCM)
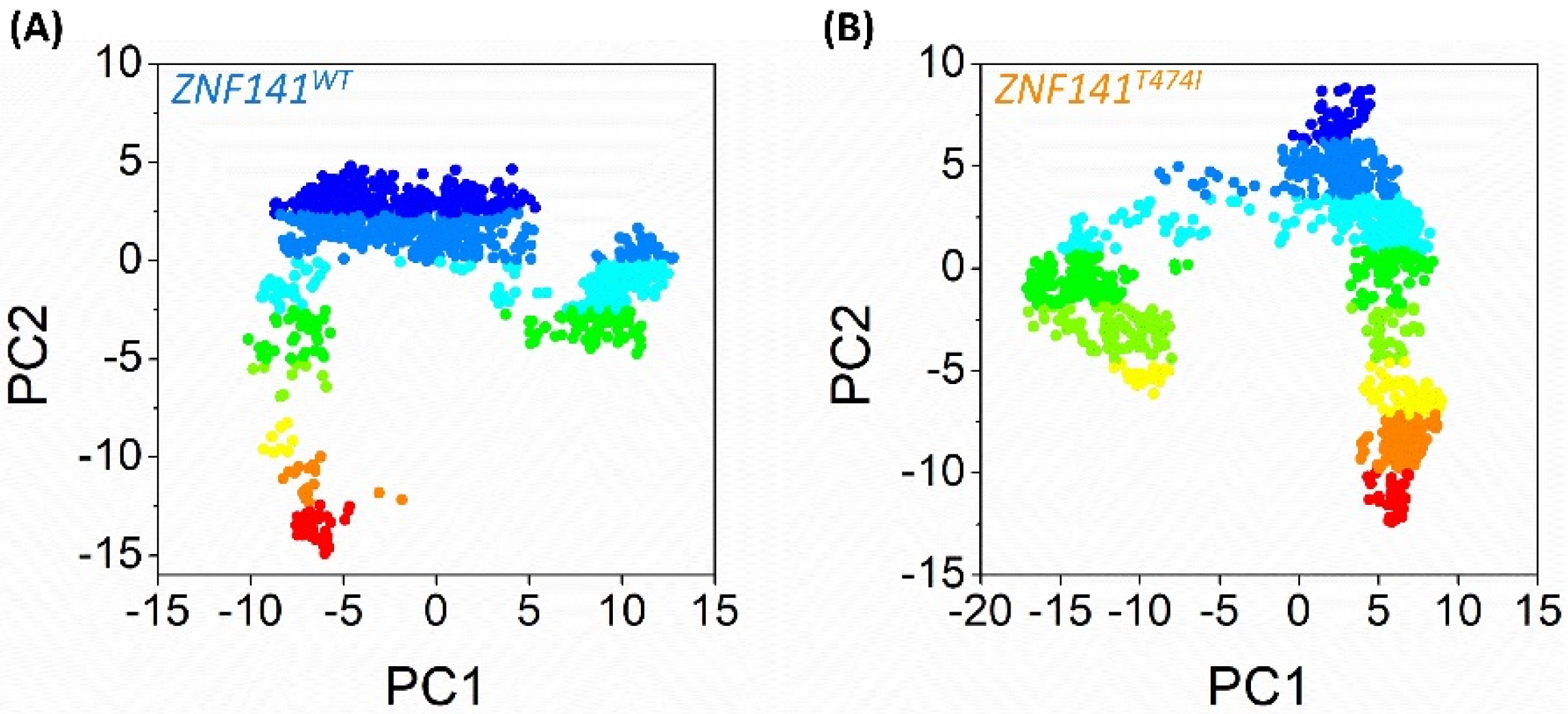
3.10. Essential Dynamics of ZNF141 Protein
3.11. Identification of Ligand, Active Sites Residues, and Enzyme Commission Number
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zhang, Z.; Sui, P.; Dong, A.; Hassell, J.; Cserjesi, P.; Chen, Y.T.; Behringer, R.R.; Sun, X. Preaxial polydactyly: Interactions among ETV, TWIST1 and HAND2 control anterior-posterior patterning of the limb. Development 2010, 137, 3417–3426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Faust, K.C.; Kimbrough, T.; Oakes, J.E.; Edmunds, J.O.; Faust, D.C. Polydactyly of the hand. Am. J. Orthop. (Belle Mead NJ) 2015, 44, E127–E134. [Google Scholar] [PubMed]
- Castilla, E.E.; Lugarinho, R.; da Graca Dutra, M.; Salgado, L.J. Associated anomalies in individuals with polydactyly. Am. J. Med. Genet. 1998, 80, 459–465. [Google Scholar] [CrossRef]
- Zguricas, J.; Bakker, W.F.; Heus, H.; Lindhout, D.; Heutink, P.; Hovius, S.E. Genetics of limb development and congenital hand malformations. Plast. Reconstr. Surg. 1998, 101, 1126–1135. [Google Scholar] [CrossRef]
- Kozin, S.H. Upper-extremity congenital anomalies. J. Bone Jt. Surg. Am. 2003, 85, 1564–1576. [Google Scholar] [CrossRef]
- Watson, B.T.; Hennrikus, W.L. Postaxial type-B polydactyly. Prevalence and treatment. J. Bone Jt. Surg. Am. 1997, 79, 65–68. [Google Scholar] [CrossRef]
- Kalsoom, U.E.; Klopocki, E.; Wasif, N.; Tariq, M.; Khan, S.; Hecht, J.; Krawitz, P.; Mundlos, S.; Ahmad, W. Whole exome sequencing identified a novel zinc-finger gene ZNF141 associated with autosomal recessive postaxial polydactyly type A. J. Med. Genet. 2013, 50, 47–53. [Google Scholar] [CrossRef] [Green Version]
- Messina, D.N.; Glasscock, J.; Gish, W.; Lovett, M. An ORFeome-based analysis of human transcription factor genes and the construction of a microarray to interrogate their expression. Genome Res. 2004, 14, 2041–2047. [Google Scholar] [CrossRef] [Green Version]
- Gray, K.A.; Yates, B.; Seal, R.L.; Wright, M.W.; Bruford, E.A. Genenames.org: The HGNC resources in 2015. Nucleic Acids Res. 2015, 43, D1079–D1085. [Google Scholar] [CrossRef]
- Zhang, W.; Xu, C.; Bian, C.; Tempel, W.; Crombet, L.; MacKenzie, F.; Min, J.; Liu, Z.; Qi, C. Crystal structure of the Cys2His2-type zinc finger domain of human DPF2. Biochem. Biophys. Res. Commun. 2011, 413, 58–61. [Google Scholar] [CrossRef]
- Nunez, N.; Clifton, M.M.K.; Funnell, A.P.W.; Artuz, C.; Hallal, S.; Quinlan, K.G.R.; Font, J.; Vandevenne, M.; Setiyaputra, S.; Pearson, R.C.M.; et al. The multi-zinc finger protein ZNF217 contacts DNA through a two-finger domain. J. Biol. Chem. 2011, 286, 38190–38201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tommerup, N.; Aagaard, L.; Lund, C.L.; Boel, E.; Baxendale, S.; Bates, G.P.; Lehrach, H.; Vissing, H. A zinc-finger gene ZNF141 mapping at 4p16.3/D4S90 is a candidate gene for the Wolf-Hirschhorn (4p-) syndrome. Hum. Mol. Genet. 1993, 2, 1571–1575. [Google Scholar] [CrossRef] [PubMed]
- Bellefroid, E.J.; Marine, J.C.; Matera, A.G.; Bourguignon, C.; Desai, T.; Healy, K.C.; Bray-Ward, P.; Martial, J.A.; Ihle, J.N.; Ward, D.C. Emergence of the ZNF91 Krüppel-associated box-containing zinc finger gene family in the last common ancestor of anthropoidea. Proc. Natl. Acad. Sci. USA 1995, 92, 10757–10761. [Google Scholar] [CrossRef] [Green Version]
- Hamilton, A.T.; Huntley, S.; Kim, J.; Branscomb, E.; Stubbs, L. Lineage-specific expansion of KRAB zinc-finger transcription factor genes: Implications for the evolution of vertebrate regulatory networks. Cold Spring Harb. Symp. Quant. Biol. 2003, 68, 131–140. [Google Scholar] [CrossRef]
- Ijaz, A.; Shah, K.; Aziz, A.; Rehman, F.U.; Ali, Y.; Tareen, A.M.; Khan, K.; Ayub, M.; Wali, A. Novel frameshift mutations in XPC gene underlie xeroderma pigmentosum in Pakistani families. Indian J. Dermatol. 2021, 66, 220. [Google Scholar] [PubMed]
- Ahmad, S.U.; Ali, Y.; Jan, Z.; Rasheed, S.; Nazir, N.u.A.; Khan, A.; Rukh Abbas, S.; Wadood, A.; Rehman, A.U. Computational screening and analysis of deleterious nsSNPs in human p 14ARF (CDKN2A gene) protein using molecular dynamic simulation approach. J. Biomol. Struct. Dyn. 2022, 1–12. [Google Scholar] [CrossRef]
- Purohit, R. Role of ELA region in auto-activation of mutant KIT receptor: A molecular dynamics simulation insight. J. Biomol. Struct. Dyn. 2014, 32, 1033–1046. [Google Scholar] [CrossRef]
- Jan, Z.; Ahmad, S.U.; Amara Qadus, Y.A.; Sajjad, W.; Rais, F.; Tanveer, S.; Khan, M.S.; Haq, I. 19. Insilico structural and functional assessment of hypothetical protein L345_13461 from Ophiophagus hannah. Pure Appl. Biol. (PAB) 2021, 10, 1109–1118. [Google Scholar]
- Khattak, S.; Rauf, M.A.; Zaman, Q.; Ali, Y.; Fatima, S.; Muhammad, P.; Li, T.; Khan, H.A.; Khan, A.A.; Ngowi, E.E. Genome-wide analysis of codon usage patterns of SARS-CoV-2 virus reveals global heterogeneity of COVID-19. Biomolecules 2021, 11, 912. [Google Scholar] [CrossRef]
- Rafique, R.; Khan, K.M.; Arshia; Kanwal; Chigurupati, S.; Wadood, A.; Rehman, A.U.; Karunanidhi, A.; Hameed, S.; Taha, M.; et al. Synthesis of new indazole based dual inhibitors of α-glucosidase and α-amylase enzymes, their in vitro, in silico and kinetics studies. Bioorg. Chem. 2020, 94, 103195. [Google Scholar] [CrossRef]
- Ajmal, A.; Ali, Y.; Khan, A.; Wadood, A.; Rehman, A.U. Identification of novel peptide inhibitors for the KRas-G12C variant to prevent oncogenic signaling. J. Biomol. Struct. Dyn. 2022, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Ali, Y.; Yasin, M.; Aziz, A.; Khan, A.W.; ur Rahman, S.; Haq, N.U. In-silico analysis of 2-cysteine peroxiredoxin genes in arabidopsis thaliana with possible role in carbon dioxide fixation through carbonic anhydrase regulation. Pak. J. Biochem. Biotechnol. 2022, 3, 175–189. [Google Scholar] [CrossRef]
- Hunt, S.E.; McLaren, W.; Gil, L.; Thormann, A.; Schuilenburg, H.; Sheppard, D.; Parton, A.; Armean, I.M.; Trevanion, S.J.; Flicek, P.; et al. Ensembl variation resources. Database 2018, 2018, bay119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sim, N.L.; Kumar, P.; Hu, J.; Henikoff, S.; Schneider, G.; Ng, P.C. SIFT web server: Predicting effects of amino acid substitutions on proteins. Nucleic Acids Res. 2012, 40, W452–W457. [Google Scholar] [CrossRef]
- Adzhubei, I.A.; Schmidt, S.; Peshkin, L.; Ramensky, V.E.; Gerasimova, A.; Bork, P.; Kondrashov, A.S.; Sunyaev, S.R. A method and server for predicting damaging missense mutations. Nat. Methods 2010, 7, 248–249. [Google Scholar] [CrossRef] [Green Version]
- Choi, Y.; Chan, A.P. PROVEAN web server: A tool to predict the functional effect of amino acid substitutions and indels. Bioinformatics 2015, 31, 2745–2747. [Google Scholar] [CrossRef] [Green Version]
- Ioannidis, N.M.; Rothstein, J.H.; Pejaver, V.; Middha, S.; McDonnell, S.K.; Baheti, S.; Musolf, A.; Li, Q.; Holzinger, E.; Karyadi, D.; et al. REVEL: An Ensemble Method for Predicting the Pathogenicity of Rare Missense Variants. Am. J. Hum. Genet. 2016, 99, 877–885. [Google Scholar] [CrossRef] [Green Version]
- Rentzsch, P.; Witten, D.; Cooper, G.M.; Shendure, J.; Kircher, M. CADD: Predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res. 2019, 47, D886–D894. [Google Scholar] [CrossRef]
- Dong, C.; Wei, P.; Jian, X.; Gibbs, R.; Boerwinkle, E.; Wang, K.; Liu, X. Comparison and integration of deleteriousness prediction methods for nonsynonymous SNVs in whole exome sequencing studies. Hum. Mol. Genet. 2015, 24, 2125–2137. [Google Scholar] [CrossRef] [Green Version]
- Hunter, S.; Apweiler, R.; Attwood, T.K.; Bairoch, A.; Bateman, A.; Binns, D.; Bork, P.; Das, U.; Daugherty, L.; Duquenne, L.; et al. InterPro: The integrative protein signature database. Nucleic Acids Res. 2009, 37, D211–D215. [Google Scholar] [CrossRef] [Green Version]
- Sali, A.; Blundell, T.L. Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 1993, 234, 779–815. [Google Scholar] [CrossRef] [PubMed]
- Ko, J.; Park, H.; Heo, L.; Seok, C. GalaxyWEB server for protein structure prediction and refinement. Nucleic Acids Res. 2012, 40, W294–W297. [Google Scholar] [CrossRef] [PubMed]
- Schmid, N.; Eichenberger, A.P.; Choutko, A.; Riniker, S.; Winger, M.; Mark, A.E.; van Gunsteren, W.F. Definition and testing of the GROMOS force-field versions 54A7 and 54B7. Eur. Biophys. J. EBJ 2011, 40, 843–856. [Google Scholar] [CrossRef] [PubMed]
- Laskowski, R.A.; MacArthur, M.W.; Moss, D.S.; Thornton, J.M. PROCHECK: A program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 1993, 26, 283–291. [Google Scholar] [CrossRef]
- Emsley, P.; Lohkamp, B.; Scott, W.G.; Cowtan, K. Features and development of Coot. Acta Crystallogr. Sect. D 2010, 66, 486–501. [Google Scholar] [CrossRef] [Green Version]
- Colovos, C.; Yeates, T.O. Verification of protein structures: Patterns of nonbonded atomic interactions. Protein Sci. A Publ. Protein Soc. 1993, 2, 1511–1519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lovell, S.C.; Davis, I.W.; Arendall, W.B., 3rd; de Bakker, P.I.; Word, J.M.; Prisant, M.G.; Richardson, J.S.; Richardson, D.C. Structure validation by Calpha geometry: Phi, psi and Cbeta deviation. Proteins 2003, 50, 437–450. [Google Scholar] [CrossRef]
- Yang, J.; Roy, A.; Zhang, Y. Protein-ligand binding site recognition using complementary binding-specific substructure comparison and sequence profile alignment. Bioinformatics 2013, 29, 2588–2595. [Google Scholar] [CrossRef] [Green Version]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [Green Version]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E.J.S. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1, 19–25. [Google Scholar] [CrossRef] [Green Version]
- Rigsby, R.E.; Parker, A.B. Using the PyMOL application to reinforce visual understanding of protein structure. Biochem. Mol. Biol. Educ. A Bimon. Publ. Int. Union Biochem. Mol. Biol. 2016, 44, 433–437. [Google Scholar] [CrossRef] [PubMed]
- Venselaar, H.; Te Beek, T.A.; Kuipers, R.K.; Hekkelman, M.L.; Vriend, G. Protein structure analysis of mutations causing inheritable diseases. An e-Science approach with life scientist friendly interfaces. BMC Bioinform. 2010, 11, 548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laskowski, R.A.; Jabłońska, J.; Pravda, L.; Vařeková, R.S.; Thornton, J.M. PDBsum: Structural summaries of PDB entries. Protein Sci. A Publ. Protein Soc. 2018, 27, 129–134. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.E.; Huang, C.C.; Ferrin, T.E. RRDistMaps: A UCSF Chimera tool for viewing and comparing protein distance maps. Bioinformatics 2015, 31, 1484–1486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, Y.; Clark, K.J.; Campbell, J.M.; Panetta, M.R.; Guo, Y.; Ekker, S.C. Making designer mutants in model organisms. Development 2014, 141, 4042–4054. [Google Scholar] [CrossRef] [Green Version]
- Segre, J.A.; Bauer, C.; Fuchs, E. Klf4 is a transcription factor required for establishing the barrier function of the skin. Nat. Genet. 1999, 22, 356–360. [Google Scholar] [CrossRef]
- Ahmad, S.U.; Hafeez Kiani, B.; Abrar, M.; Jan, Z.; Zafar, I.; Ali, Y.; Alanazi, A.M.; Malik, A.; Rather, M.A.; Ahmad, A.; et al. A comprehensive genomic study, mutation screening, phylogenetic and statistical analysis of SARS-CoV-2 and its variant omicron among different countries. J. Infect. Public Health 2022, 15, 878–891. [Google Scholar] [CrossRef]
- Shah, A.A.; Amjad, M.; Hassan, J.-U.; Ullah, A.; Mahmood, A.; Deng, H.; Ali, Y.; Gul, F.; Xia, K. Molecular Insights into the Role of Pathogenic nsSNPs in GRIN2B Gene Provoking Neurodevelopmental Disorders. Genes 2022, 13, 1332. [Google Scholar] [CrossRef]
- Wadood, A.; Shareef, A.; Ur Rehman, A.; Muhammad, S.; Khurshid, B.; Khan, R.S.; Shams, S.; Afridi, S.G. In Silico Drug Designing for ala438 Deleted Ribosomal Protein S1 (RpsA) on the Basis of the Active Compound Zrl15. ACS Omega 2022, 7, 397–408. [Google Scholar] [CrossRef]
- Khan, M.S.; Mehmood, B.; Yousafi, Q.; Bibi, S.; Fazal, S.; Saleem, S.; Sajid, M.W.; Ihsan, A.; Azhar, M.; Kamal, M.A. Molecular docking studies reveal rhein from rhubarb (rheum rhabarbarum) as a putative inhibitor of ATP-binding cassette super-family G member 2. Med. Chem. 2021, 17, 273–288. [Google Scholar] [CrossRef]
- Essadssi, S.; Krami, A.M.; Elkhattabi, L.; Elkarhat, Z.; Amalou, G.; Abdelghaffar, H.; Rouba, H.; Barakat, A. Computational Analysis of nsSNPs of ADA Gene in Severe Combined Immunodeficiency Using Molecular Modeling and Dynamics Simulation. J. Immunol. Res. 2019, 2019, 5902391. [Google Scholar] [CrossRef] [PubMed]
- Yue, P.; Moult, J. Identification and analysis of deleterious human SNPs. J. Mol. Biol. 2006, 356, 1263–1274. [Google Scholar] [CrossRef] [PubMed]
- Bibi, S.; Sakata, K. An integrated computational approach for plant-based protein tyrosine phosphatase non-receptor type 1 inhibitors. Curr. Comput.-Aided Drug Des. 2017, 13, 319–335. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| S.No. | Protein | RMSD (nm) (Average) | RMSF (nm) (Average) | RMSF (T474I) | H-Bonds (Average) | Rg (nm) | SASA (nm2) (Average) |
|---|---|---|---|---|---|---|---|
| 1 | wt-ZNF141 | 1.71 | 0.91 | 1.17 | 207 | 2.9 | 226.19 |
| 2 | T474I | 1.76 | 1.11 | 2.39 | 200 | 3.4 | 235.66 |
| Rank | C-Score | Cluster Size | PDB Hit | Lig Name | Binding Residues |
|---|---|---|---|---|---|
| 1 | 0.17 | 14 | 2lt7A | ZN | 257,260,273,277 |
| 2 | 0.05 | 6 | 1a1iA | Nuc.acid | 337,346,348,350,353,356,357,360,374,376,377, 378,381,385,388,402,406,409,412 |
| 3 | 0.04 | 4 | 3g0bD | NAG | 229,232,236,245 |
| 4 | 0.04 | 3 | 2wbsA | ZN | 285,288,301,305 |
| 5 | 0.03 | 3 | 2i13B | Nuc.acid | 337,346,348,350,353,356,357,374,376,377,378, 381,385,388,402,404,406,409,412,413,416 |
| 6 | 0.03 | 3 | 1tf6A | Nuc.acid | 262,264,265,266,269,273,276,292,296,297,300, 304,318,320,322,325,329,332,348,350,353,357, 360,376,378,381,385,388,391 |
| 7 | 0.02 | 2 | 4kx7A | ZN | 313,315,316,329,334 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ali, Y.; Ahmad, F.; Ullah, M.F.; Haq, N.U.; Haq, M.I.U.; Aziz, A.; Zouidi, F.; Khan, M.I.; Eldin, S.M. Structural Evaluation and Conformational Dynamics of ZNF141T474I Mutation Provoking Postaxial Polydactyly Type A. Bioengineering 2022, 9, 749. https://doi.org/10.3390/bioengineering9120749
Ali Y, Ahmad F, Ullah MF, Haq NU, Haq MIU, Aziz A, Zouidi F, Khan MI, Eldin SM. Structural Evaluation and Conformational Dynamics of ZNF141T474I Mutation Provoking Postaxial Polydactyly Type A. Bioengineering. 2022; 9(12):749. https://doi.org/10.3390/bioengineering9120749
Chicago/Turabian StyleAli, Yasir, Faisal Ahmad, Muhammad Farhat Ullah, Noor Ul Haq, M. Inam Ul Haq, Abdul Aziz, Ferjeni Zouidi, M. Ijaz Khan, and Sayed M. Eldin. 2022. "Structural Evaluation and Conformational Dynamics of ZNF141T474I Mutation Provoking Postaxial Polydactyly Type A" Bioengineering 9, no. 12: 749. https://doi.org/10.3390/bioengineering9120749