1. Introduction
The bone marrow niche is defined as a local tissue microenvironment that maintains and regulates hematopoietic stem cells (HSCs). Many previous studies have suggested that mesenchymal stem cells (MSCs) and vascular endothelial cells maintain HSCs and regulate their proliferation and differentiation in the bone marrow niche [
1,
2,
3,
4]. The bone marrow niche has been rapidly elucidated in recent years by next-generation sequencing and cell analysis techniques [
5].
In order to analyze the bone marrow niche, many studies have attempted to develop humanized hematopoietic models, such as patient-derived xenograft (PDX) and humanized ossicle (hOss), which are small human bone organs with structures and functions similar to mouse bones [
6,
7]. PDX models need to collect patient-derived cells and have difficulty preparing cells with genetically equivalent conditions. HOss models require bone marrow-derived MSCs (BM-MSCs) to differentiate osteogenic lineages and be seeded onto scaffold material prior to the implantation of HSCs.
In recent years, various biomaterials have been generated by the recellularization of cells into the skeleton of decellularized extracellular matrices (dECM) [
8]. We focused on porcine decellularized bone (DCB) as a scaffold, which is categorized as a dECM. Hashimoto et al. reported that the subcutaneous transplantation of DCBs into rats established hematopoietic tissue in DCBs without provoking an immune response [
9]. According to Nakamura et al., sodium-dodecyl-sulfate (SDS)-treated DCB, stripped of reticular and adipose tissues, lacks long-term HSCs in vivo, which can be found in high hydrostatic pressure-treated DCBs, which were used in our study [
10]. Unlike the hOss model, there is no need to wait for seeded BM-MSCs to differentiate into osteogenic lineages. DCB with reticular and adipose tissue is the scaffold of choice to create a microenvironment for hematopoiesis.
Myelodysplastic syndrome (MDS) is a hematological neoplasm in which abnormal HSCs with genetic mutations proliferate. Recent studies have reported that more MSCs are in contact with HSCs in MDS [
11]. Genetic abnormalities, such as the
DICER1 mutation in MSCs, not HSCs, may induce MDS [
11,
12,
13]. Mian et al. cultured MDS patient-derived HSCs and MSCs in gelatin sponge, transplanted them into mice, and showed a sustained human MDS niche in vivo [
14]. Patient-derived samples are heterogeneous for each patient. To analyze the disease onset of human MDS in the bone marrow niche, it is necessary to prepare hMSCs with identical genetic abnormalities, induct them to certain scaffolds, and transplant them to mice. We hypothesize that DCB is an appropriate biomaterial because it enables the analysis of the interactions of hMSCs and mouse hematopoietic cells without adding any cytokines or differentiation factors.
We hypothesized that if genetically engineered human MSCs (hMSCs) were introduced and grown on DCBs, the subcutaneous implantation of these DCBs in mice could recapitulate the human–mouse hybrid bone marrow niche that could support the interaction of hMSCs and mouse hematopoietic cells.
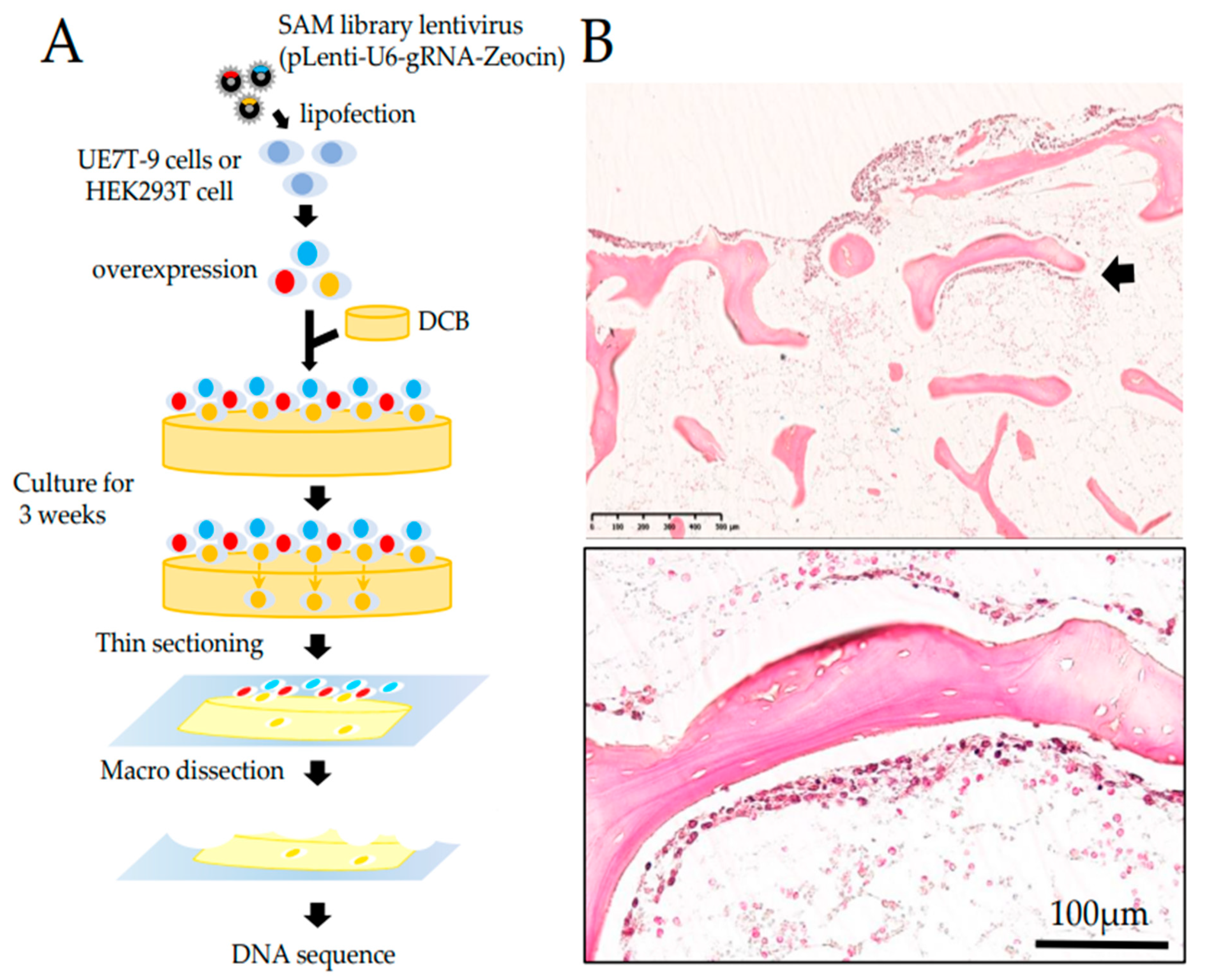
The induction of hMSCs into DCB was difficult. We first cultured UE7T-9 cells (human bone marrow-derived MSC cell line) on DCB, but failed to achieve intraosseous expansion (
Supplementary Figure S1A,B). Nakamura et al. demonstrated that culturing human bone marrow stem cells with DCB at 4 °C reduced their cell adhesive capacity and allowed them to infiltrate the reticular tissue within DCB in the short term. However, the distance of cell infiltration decreases with the days in culture, suggesting that the long-term engraftment of human bone marrow stem cells into DCB is very difficult [
15]. Introducing hMSCs into DCBs is the very important first step toward establishing a hybrid bone marrow niche in vivo.
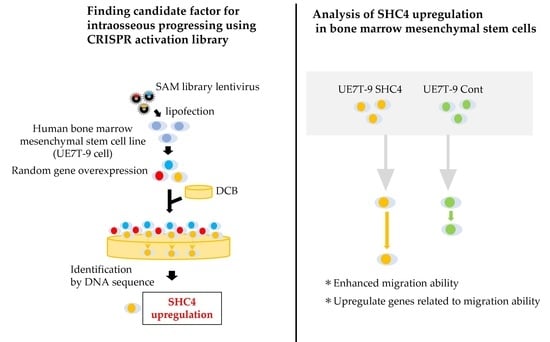
We used the clustered, regularly interspaced short palindromic repeats Synergistic Activation Mediator library (CRISPR SAM library). The CRISPR library is a gene-editing technology that has been used for research worldwide [
16,
17]. The CRISPR SAM library enables the overexpression of target genes using dCas9 (Cas9 with the catalytic domain deactivated) and MS2-p65-HSF1 protein [
18]. Using this system, the CRISPR SAM library can upregulate genes randomly induced by sgRNAs and identify the genes by sequencing the selected sgRNA after selection.
To recapitulate the infiltration of hMSCs into DCB, we sought to identify the factors that promote the progression of UE7T-9 cells into DCB using the CRISPR SAM Library. We validated the identified genes that would promote UE7T-9 cell integration in DCB.
2. Materials and Methods
2.1. Cell lines and Cell Culture
Human mesenchymal stem cells (UE7T-9 cells) were purchased from the Japanese Cancer Research Bank (JCRB, Osaka, Japan) cell bank in 2014 (
https://cellbank.nibiohn.go.jp/~cellbank/cgi-bin/search_res_det.cgi?ID=3821, accessed on 2 May 2022). Human embryonic kidney cells (HEK 293T) were also purchased from JCRB. UE7T-9 cells and HEK 293T cells were maintained with Dulbecco’s modified Eagle’s medium (D-MEM, Fujifilm) supplemented with 10% fetal bovine serum (Thermo Fisher Scientific, Walthum, MA, USA) and penicillin/streptomycin (Thermo Fisher Scientific, Walthum, MA, USA), incubated at 37 °C with 5% CO
2. D-MEM was used as the culture medium throughout the experiments.
2.2. Transfection of SAM Library into Cell Lines
To transduce the lentivirus, HEK 293T cells (5 × 10
5 cells/well) were seeded in a six-well cell culture plate for one day before transfection. The next day, 3 µg of lentivirus plasmid was transfected with 1 µg of pMD2.G and 2 µg of pCMV using Lipofectamine™ 3000 Reagent (Thermo Fisher Scientific, Walthum, MA, USA), according to previous work [
19]. Twelve hours after transfection, the medium was changed to fresh DMEM. The virus supernatant was harvested 48 h after transfection and then filtered with Millex™ -HP 0.45 µm (Merck, Kenilworth, NJ, USA). The target HEK 293T cells (5 × 10
5 cells/well) and UE7T-9 cells (2 × 10
5 cells/well) were seeded in a six-well cell culture plate one day prior to transduction. The cells were transduced with this lentivirus supernatant with 5 µg/mL polybrene (Sigma-Aldrich, MO, USA). Transduction was performed using spin infection followed by 30 min of centrifugation at 4680 rpm. The lentiviral plasmids were lentiMPHv2 (Addgene, #89308, MA, USA), lenti-dCAS-VP64_blast (Addgene, #61425), and lentiSAMv2 (Addgene, #61597) [
17]. First, lentiMPHv2 was transduced into HEK 293T and UE7T-9 cells, and transduced cells were selected using hygromycin B for 2 weeks. Then, lenti-dCAS9-VP64 was induced in MPH-expressing HEK 293T cells and UE7T-9 cells, and selected by Blasticidin (10 μg/mL, Thermo Fisher Scientific, Walthum, MA, USA) for 2 weeks. MPH-dCas9-VP64-expressing HEK 293T and UE7T-9 cells were applied to further experiments. The CRISPR-activated library, lenti-SAMv2, was also transduced into MPH-dCas9-VP64-expressing UE7T-9 cells. Although the multiplicity of infection (MOI) is usually set to 1 or less, in this study, we allowed duplicate infections (MOI > 1) because our goal was to invade DCB. We then performed selections with zeocin (300 μg/mL, Thermo Fisher Scientific, Walthum, MA, USA) for 2 weeks.
2.3. DCB Formation
Bone preparation was performed as described in a previous report [
8,
13]. Fresh pig femurs and ribs were obtained from a local slaughterhouse (Tokyo Shibaura Shipbuilding, Tokyo, Japan). Femurs (compact bones) and costae (trabecular bones) were cut into small pieces (4 × 4 × 3 mm) and washed in phosphate buffered saline (PBS) (Invitrogen, Tokyo, Japan) containing penicillin (100 units/mL) and streptomycin (100 μg/mL). The bone/marrow was filled with PBS and sealed in a plastic pack to prevent implosion and leakage during the pressure application. The cells were dissociated by hydrostatic pressurization at 980 MPa and 30 °C for 10 min using a cold isostatic pressurizer (Dr. CHEF, Kobe Steel, Ltd., Hyogo, Japan). Pressurization and decompression were performed at 65.3 MPa/min, and propylene glycol was used as the permeate. The cells were then washed with saline containing DNase I (0.2 mg/mL) (Roche Diagnostics, Tokyo, Japan) antibiotic for 3 weeks at 37 °C with continuous slow shaking, and then treated with 80% v/v EtOH at 37 °C for 7 days. After washing and shaking with PBS again, they were stored at 4 °C.
2.4. Cell Culture on DCB and Histological Evaluation
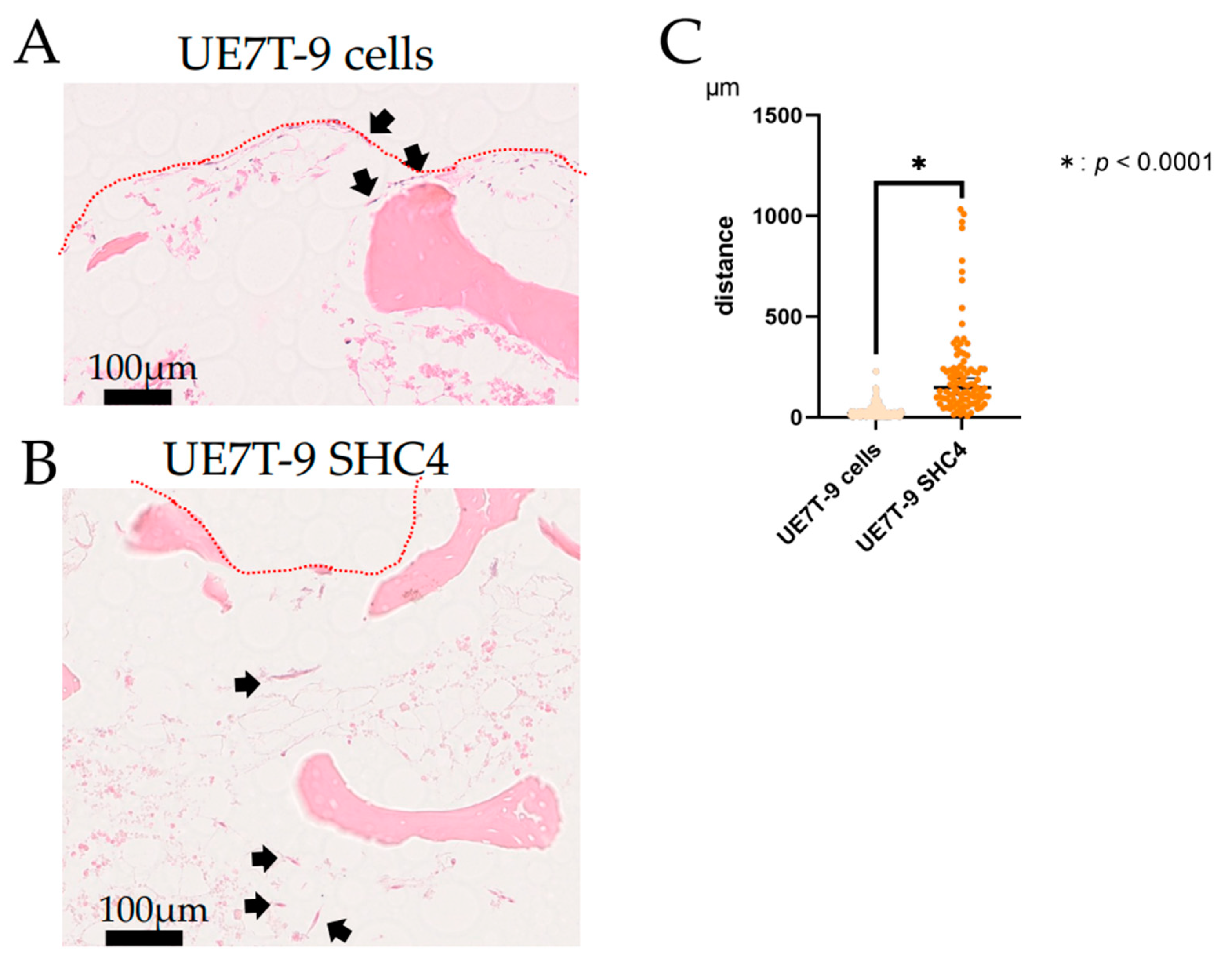
To evaluate infiltration into DCB, HEK 293T cells and UE7T-9 cells (1.0 × 10⁵ cells/well) were cultured on DCB for 3 or 8 weeks. After culture, the cells were fixed in 10% neutral buffered formalin for 1 day and demineralized in 18.5% EDTA solution (Pharma, Tokyo, Japan) for 1 day. Then paraffin-embedded sections were prepared, and hematoxylin-eosin staining of thin sections (3 μm) was performed. We photographed stained sections with a NanoZoomer S210 virtual slide scanner (Hamamatsu Photonics, Shizuoka, Japan) and measured distances using NDP.view2 (Hamamatsu Photonics K.K., Hamamatsu, Japan).
2.5. DNA Sequence Analysis
Paraffin-embedded sections prepared as described in the section “Cell culture on DCB and histological evaluation” were thinly sliced at 10 μm. For five sections of DCB with surface cells removed, we added 0.5 mL of DEXPAT (TaKaRa, Shiga, Japan), heated at 100 °C for 10 min, and centrifuged at 4 °C at 12,000 rpm for 10 min. The aqueous layer portion was collected, avoiding the paraffin thin film, and then purified with Wizard SV Gel & PCR Clean-up System (Promega, Madison, WI, USA ). For gRNA detection, PCR by the KOD-FX (TOYOBO, Osaka, Japan) method was then performed using SAM 6025F (tttcttgggtagtttgcagt) and SAM 6190R (cctcatgttggcctagctctct) as primers. The PCR products were purified and introduced into Zero Blunt TOPO vector using Zero Blunt TOPO PCR Cloning Kit (Thermo Fisher Scientific, Walthum, MA, USA). We performed DNA sequence analysis (Sanger method), identified gRNA, and searched for the corresponding gene in the gRNA list of the CRISPR SAM Library.
2.6. Creation of SHC4-Overexpressing Cell Lines
The gene fragments for the coding region of
SHC4 with SAL1 and HIND3 restriction enzyme sites at each end were purchased from Integrated DNA Technologies (
Supplementary Figure S2 for sequence diagram), integrated into 150 ng pENTR221 vectors, and incubated at room temperature for 4 h. pENTR221-GFP was used as a control. Next, it was replaced with 150 ng of plenti CMV Puro DEST vector (Plasmid #17452, Addgene) by LR reaction usingGateway
® LR Clonase (Thermo Fisher Scientific, Walthum, MA, USA) and incubated at room temperature for 4 h. We collected the plasmid using Wizard
® Plus SV Minipreps DNA Purification Systems (Promega, Madison, WI, USA) and confirmed the presence of the target bands with electrophoresis. We created two types of plasmids (plenti CMV GFP puro and plenti CMV
SHC4 puro). We then performed lipofection as described in the section “Transfection of SAM library into cell lines”, and we collected viral supernatant, which was used to infect UE7T-9 cells (2.0 × 10⁵ cells/well each). We adjusted the culture volume to 25 μg/mL/well, and then performed selection using puromycin.
2.7. Western Blot
We used an SDS buffer (25% of 0.125M Tris-HCl (pH 6.8), 20% of glycerol, 4% of SDS, and 10% of 2-mercaptoethanol, with bromophenol blue included) to pellet cell pellets, extract proteins, and degrade DNA by sonication in ice-cold conditions. The protein extracts were separated in Mini-PROTEAN TGX Stain-Free gels (BIO RAD, CA, USA) by electrophoresis (60 mA, 40 min) and transferred to TransBlot Turbo Mini-size PVDF membranes (BIO RAD). The membranes were blocked with Bullet Blocking One (Nacalai Tesque, Kyoto, Japan). The antibodies we used were as follows: primary antibodies—SHC4 polyclonal antibody (rabbit, 75 kDa) (Sigma-Aldrich, Mizulli, USA) and β-actin monoclonal antibody (rabbit; 45 kDa) (Cell signaling technology, #4790, MA, USA) diluted 1000×, reacted at 4 °C in overnight. The membranes were washed with TBS-T, and the horseradish peroxidase (HRP)-conjugated anti-rabbit antibody (GE Healthcare, Chicago, IL, USA) diluted 5000× was reacted with membrane for 1 h. The membranes were washed with TBS-T three times before being exposed to the ClarityTM Western ECL Substrate (Bio Rad Laboratories, Hercules, CA, USA) as a chromogenic substrate for HRP. The protein bands were detected using a ChemiDoc MP ImageLab (Bio Rad Laboratories, Hercules, CA, USA).
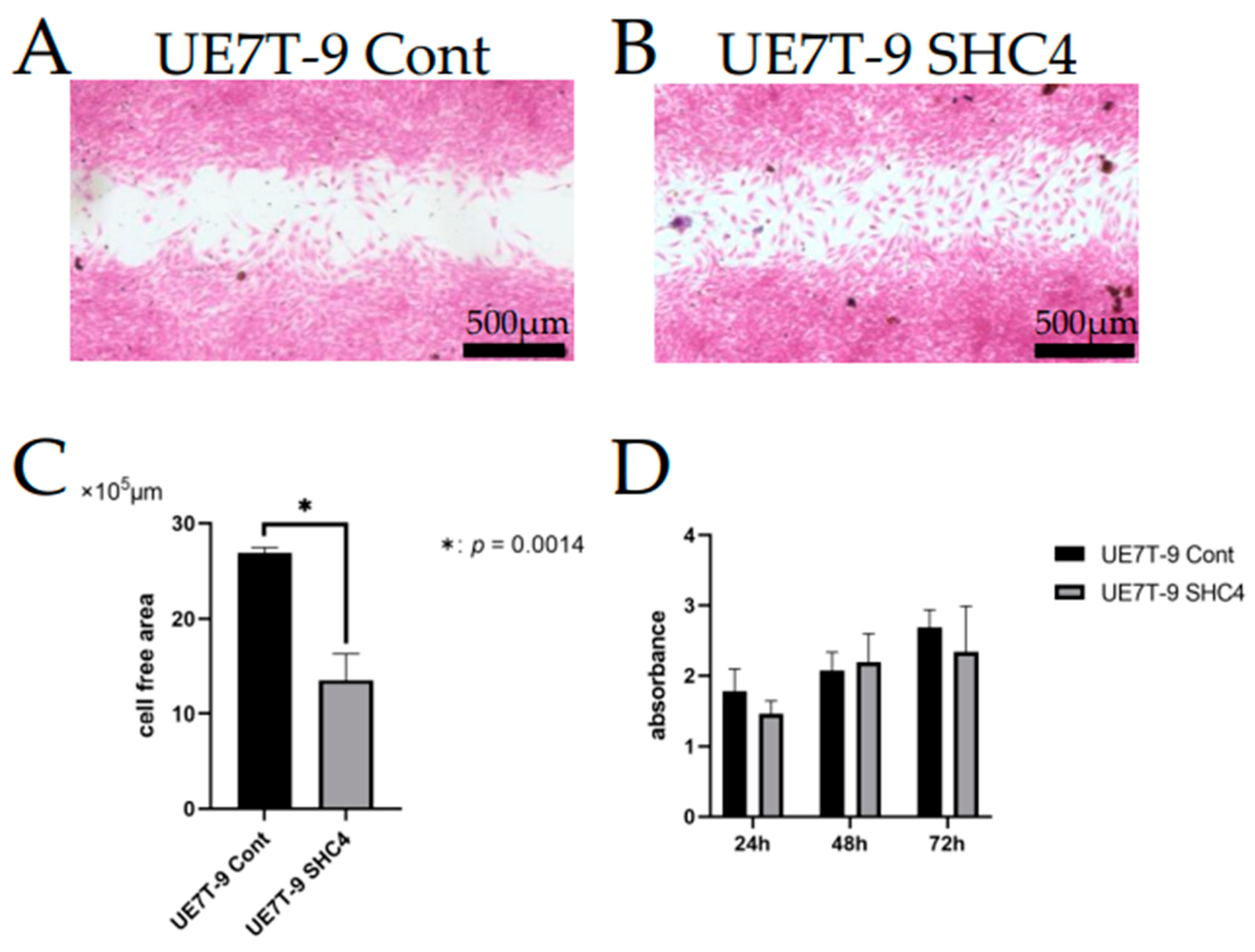
2.8. MTS Assay
An MTS assay was performed to evaluate cell proliferation. We cultured UE7T-9 cells at 0.5 × 10⁴ cells/well in 96-well plates. At 24, 48, and 72 h of culture, we added 20 µL of MTS (2 mg/mL) (Promega, Madison, WI, USA )/PMS (0.92 mg/mL) (Promega, Madison, WI, USA) to each well. The wells were light-shielded and placed in an incubator for 2–3 h. We measured absorbance at 490 nm and 650 nm using a microplate reader ELx808 IU BioTek Instruments Inc. (Thermo Fisher Scientific, Walthum, MA, USA). Data were statistically analyzed using the Student’s t-test.
2.9. Scratch Assay
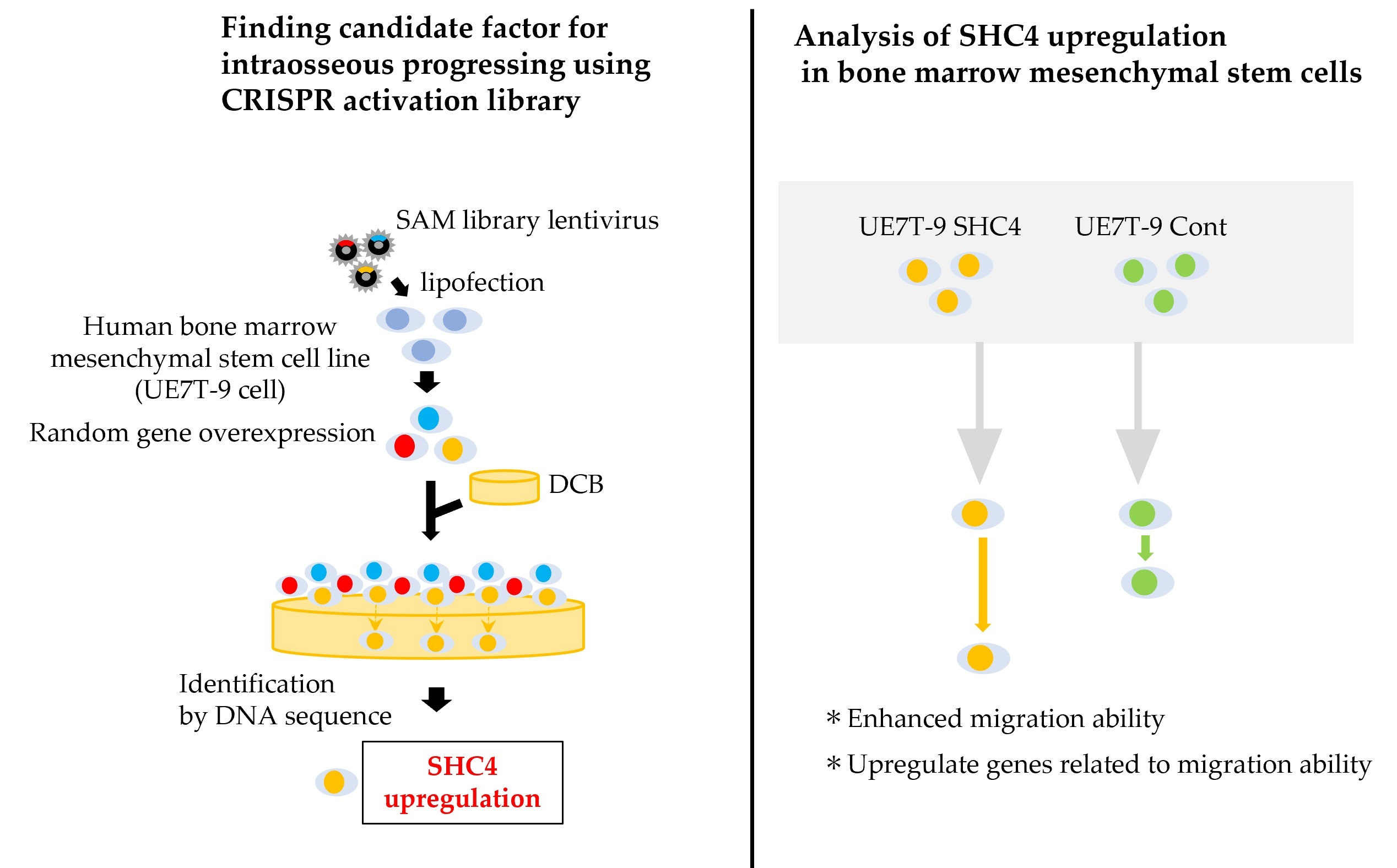
We used a scratch assay to evaluate cell migration ability. UE7T-9 cells were cultured at 3.0 × 10⁴ cells/70 µL using culture inserts (Ibidi, Gräfelfing, Germany). After 24 h, we removed the culture inserts, and HEK293T cells were cultured for 18 h. Formalin fixation and HE staining were then performed. The area between inserts after cell migration (cell-free area) was measured using Image J 1.53k (Wayne Rasband and contributors, National Institutes of Health, MD, USA). Data were statistically analyzed using the Student’s t-test.
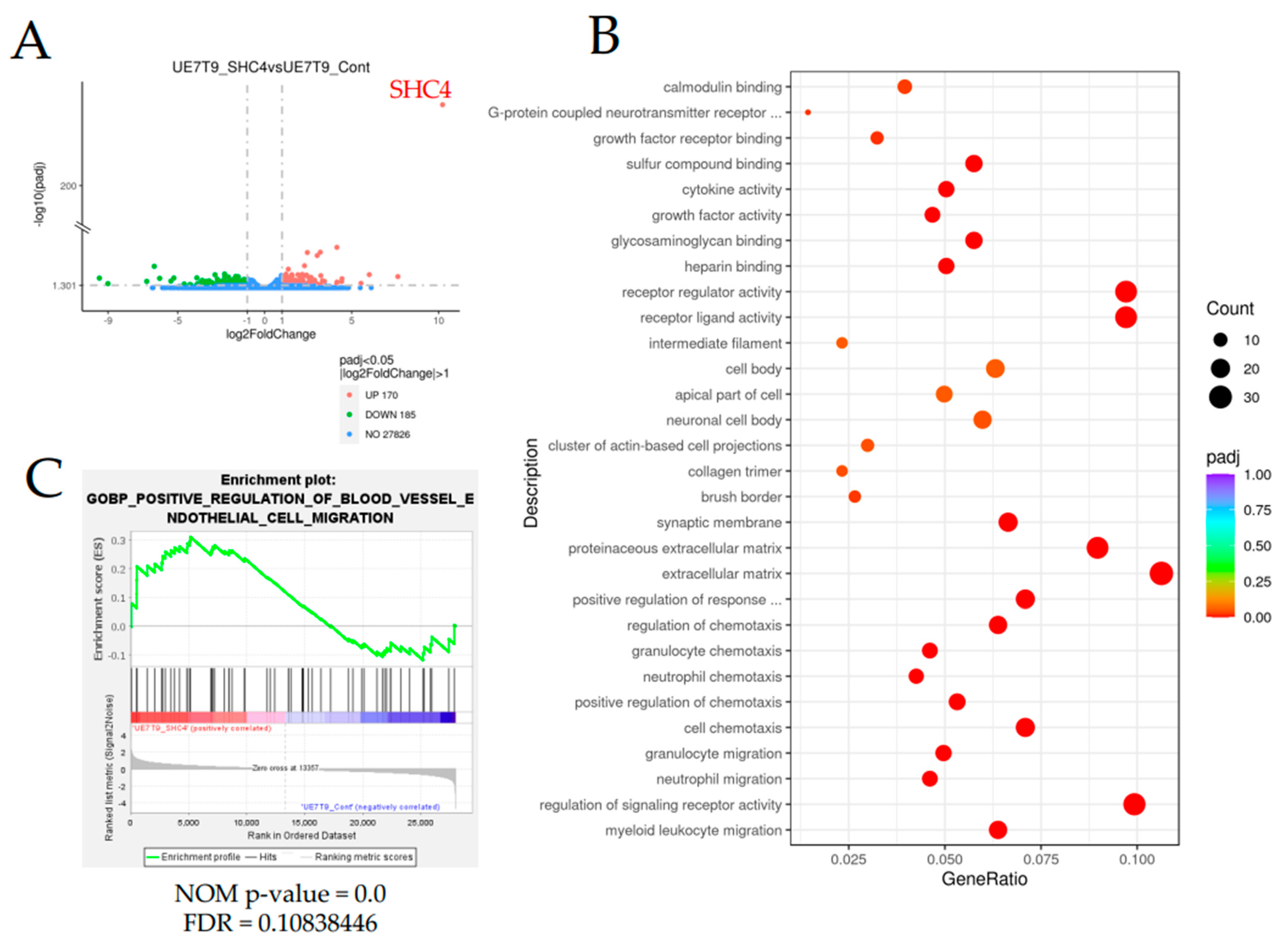
2.10. Description of RNA Sequence Analysis
Total RNA was extracted from three samples, each of UE7T-9 SHC4 and UE7T-9 Cont, using a RNeasy Mini Kit (QIAGEN, Hilden, Germany). All library prep and whole genome sequencing were performed by Novogene Japan K.K. (Japan,
https://jp.novogene.com/, accessed on 12 July 2022) using the NovaSeq 6000 Sequencing System (Illumina, CA, USA). Volcano plots were generated using Novosmart software (Novogene). Enrichment analysis was performed using clusterProfiler (Yu G, 2012) software based on Gene Ontology (GO), Disease Ontology (DO), Kyoto Encyclopedia of Genes and Genomes (KEGG), and Reactome data. For GSEA, normalized expression data were analyzed and visualized with GSEA software (version 4.2.3,
https://www.gsea-msigdb.org/gsea/index.jsp, accessed on 27 May 2022). The normalized enrichment score (NES), nominal p-value (NOM-p), and false discovery rate q-value (FDR-q) were calculated for comparison, and the categories selected were universally upregulated in UE7T-9 cells. The relative enrichment of individual genes was assessed based on the rank metric score following the GSEA.
2.11. Statistical Analysis
The data were statistically analyzed using Graph Pad Prism Version 9.3.1 (350) software. The results were expressed as the mean ± standard deviation (SD). Statistical significance was determined by the Student’s t-test. A value of p < 0.05 was considered statistically significant for all analyses.
4. Discussion
The introduction of hMSCs into DCB, a biomaterial, is very important for reconstructing the hybrid bone marrow niche. Despite various efforts in the past, the introduction of these cell lines has been difficult. Previous studies that have attempted to create models of the bone marrow niche have used primary hMSCs [
12]. UE7T-9 cells are immortalized primary hMSCs introducing telomerase reverse transcriptase (hTERT) genes and human papillomavirus E7 genes. UE7T-9 cells are proliferative and can be easily transfected with lentiviral vectors. Further studies using other mesenchymal cell lines and primary hMSCs are necessary. In this study, we succeeded in introducing a human bone marrow mesenchymal cell line into DCB by using the CRISPR activation library. Notably, only
SHC4 was identified as a candidate gene for the infiltration of cells into DCB. Thus, we believe that CRISPR-activated libraries have high potential for application to the bone marrow niche, such as the introduction of cells into biomaterials, by supporting the easy and comprehensive upregulation of genes. Attempts to combine biomaterial with CRISPR for applications in disease modeling and regenerative medicine have begun to emerge in recent years [
20,
21], and this is the first study to apply the CRISPR library to dECM.
SHC4 is a member of the
Src gene family;
Src family proteins function as adapter molecules for phosphotyrosine in a variety of receptor-mediated signaling pathways. The SHC family encodes at least six Shc-like proteins, characterized by an NH2-terminal phosphotyrosine-binding (PTB) domain, a central region rich in proline and glycine residues (CH1), and a COOH-terminal Src homologue 2 domain (SH2), in the presented order.
SHC4 is located on human chromosomes on 15q21.1–q21.2 and is specifically expressed during embryonic stem cell differentiation and early embryogenesis [
22].
SHC4 is expressed at high levels in adult brain tissue and at low levels in skeletal muscle (
https://www.proteinatlas.org/ENSG00000185634-SHC4/tissue, accessed on 21 June 2022). In 2007, Ernesta Fagiani et al. identified
SHC4 (called RaLP at first) in vertically invading and metastasizing melanomas and showed that
SHC4 behaves as a substrate for IGF-1R or EGFR to promote melanoma cell migration [
22]. Jones et al. showed that
SHC4 mediates signaling downstream of phosphorylated muscle-specific kinase (MuSK) receptor tyrosine kinase at the neuromuscular junction [
23]. Melanie et al. found that
SHC4 promoted the autophosphorylation of EGFR in the absence of external stimuli. The authors used a quantitative PCR (qPCR) analysis of biopsy cores representing Grade I–IV astrocytomas to demonstrate the significantly higher expression of
SHC4 in tumor tissue compared to benign tissue [
24,
25]. Xin Zhang et al. showed that
SHC4 is expressed in hepatocellular carcinoma tissues and that
SHC4 promotes hepatocellular carcinoma progression by activating STAT3 signaling. They further noted that a high expression of
SHC4 is associated with clinicopathological features and poor prognoses in patients with hepatocellular carcinoma [
24].
SHC4 works as an oncogene and causes carcinoma cells to proliferate and become malignant. However, there have been few reports on
SHC4, and its detailed function and how it works in normal cells, especially bone marrow mesenchymal cells, remain to be elucidated.
We evaluated the effect of
SHC4 overexpression on UE7T-9 cells. We found that
SHC4 overexpression did not promote cell proliferation but significantly enhanced cell migration in UE7T-9 cells. This was confirmed by the RNA sequence analysis, which showed an enrichment of genes involved in the pathway of cell migration in UE7T-9 SHC4 compared to the control, suggesting that
SHC4 overexpression enhanced the expression of genes that promote the migratory ability of UE7T-9 cells. We hypothesized that
SHC4 overexpression in UE7T-9 cells, inducing STAT5A upregulation, may lead to MSC migration [
26].
SHC4 upregulation in UE7T-9 cells has a minimal effect on cell morphology or proliferative potential. Given that SHC4 does not involve major alterations in the properties of human bone marrow mesenchymal cells, it is a promising candidate for studying MDS caused by other genetic abnormalities, such as DICER1, in human mesenchymal cells. However, cell infiltration into DCB with SHC4 overexpression was limited to DCB. In vivo transplantation experiments require a deeper penetration of cells into the bone marrow matrix. In the future, we will consider using a third-generation CRISPR activation library, such as the Calabrese library, or using the CRISPR knockout library to search for other infiltrating factors. We believe that the successful infiltration and engraftment of hMSCs into DCB will provide a milestone for recapitulating the bone marrow niche. If the human–mouse hybrid bone marrow niche can be reproduced in mice, it will be easier to create models of MDS by inducing specific genetic abnormalities, such as DICER1, in hMSCs. Transplanted DCBs are easy to recollect and analyze the tumorigenesis of MDS for each time course.


 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}



