
WinBEST-KIT: Biochemical Reaction Simulator for Analyzing Multi-Layered Metabolic Pathways


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Overview of WinBEST-KIT
- Reaction schemes can be created by operations that are similar to using a drawing tool.A reaction scheme representing metabolic pathways can be created by connecting reactant symbols that indicate reactants and reaction step symbols that indicate reaction steps with lines. A wide variety of editing functions (cut, copy, paste, undo, and so on) are also provided;
- Reaction schemes can be created that contain a combination of mass action law and steady-state approximations of enzyme kinetics.Reaction schemes can be created that combine detailed models based on the mass action law along with the steady-state approximations of enzyme kinetics such as the Michaelis–Menten equation. The major steady-state approximations of enzyme kinetics are provided as standard reaction step symbols;
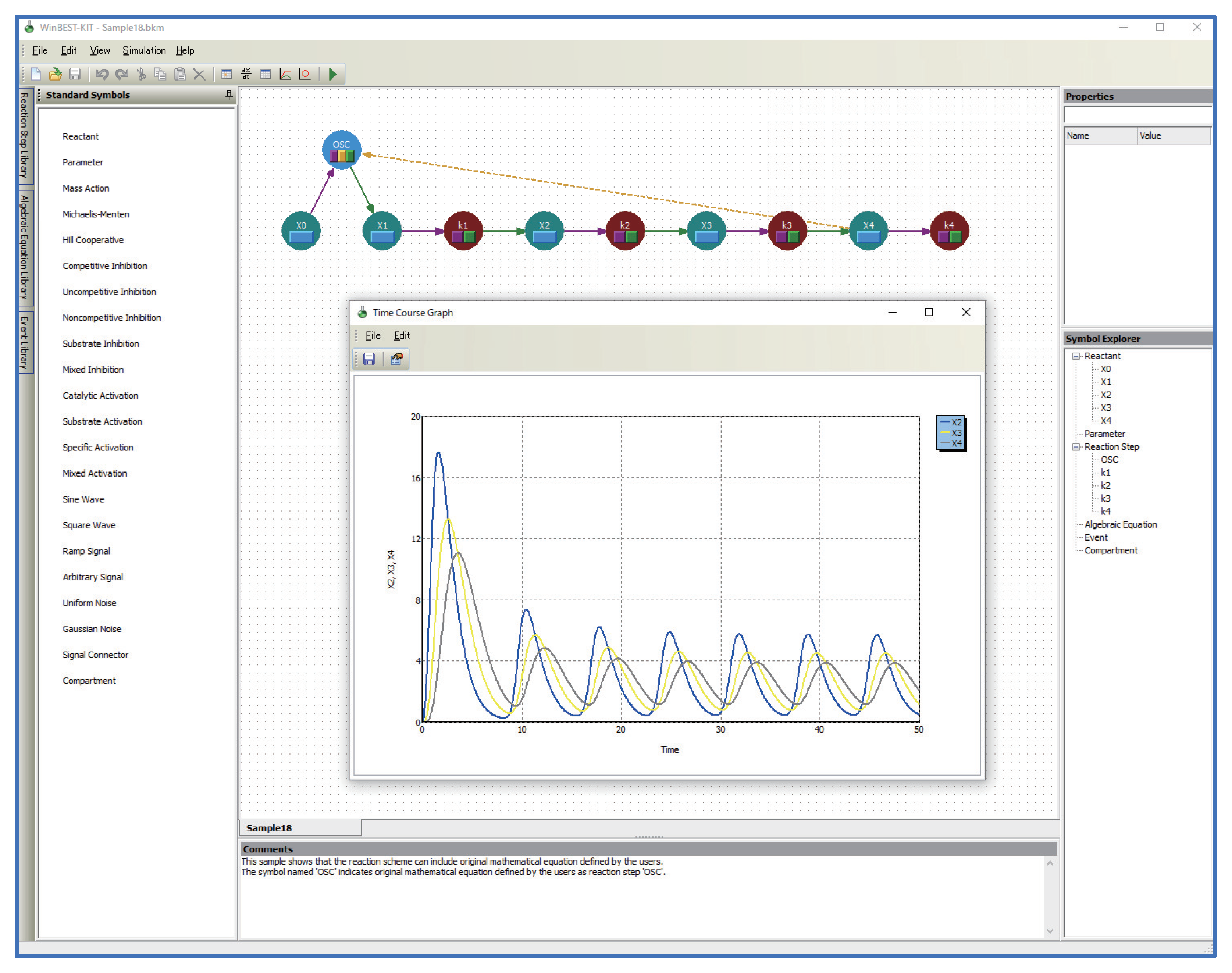
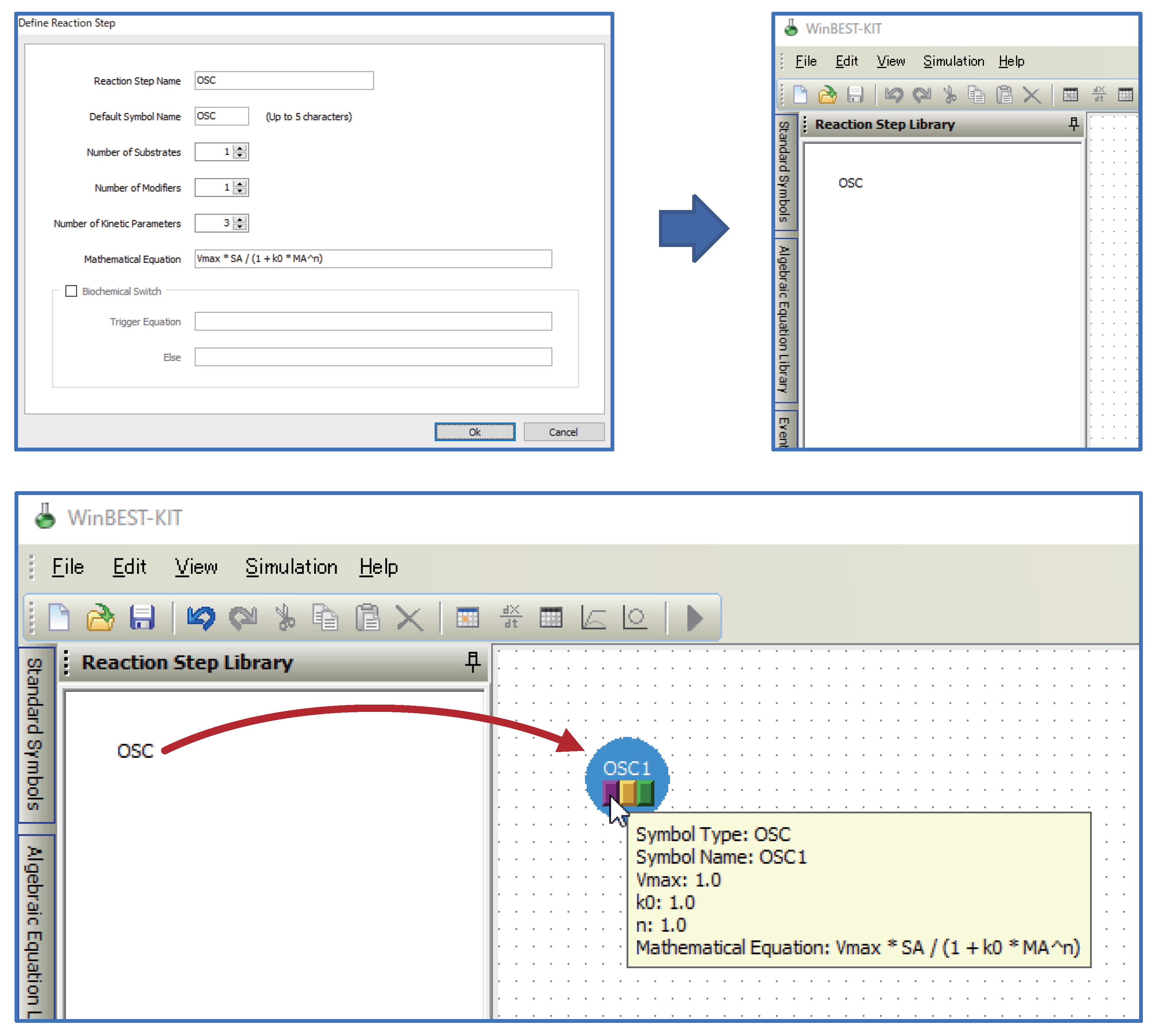
- Kinetic equations can be used as user-defined symbols for representing unknown kinetic mechanisms.Users can define kinetic equations as user-defined symbols and customize them into the diagrammed modeling interface; that is, the custom reaction step symbols can be used in WinBEST-KIT. This feature is called “Reaction Step Library.” Figure 2 shows an example of defining and customizing the kinetic equations. In Figure 2, a kinetic equation labeled “OSC” is defined and customized into the symbol selection area; thus, users can visually arrange any number of “OSC” reaction step symbols onto the editing area anytime and anywhere. Furthermore, the kinetic equations can also be approximated a part of metabolic pathways. Thus, “Reaction Step Library” can also be used as compartmentalization of biochemical reactions;
- Algebraic equations (AssignmentRule) and events, the characteristic features of SBML, can be used as user-defined symbols.Users can define algebraic equations and events as user-defined symbols and customize them into the diagrammed modeling interface through the same interface as used in the definition of the kinetic equations shown in Figure 2; that is, the custom algebraic equation symbols and the custom event symbols can be used in WinBEST-KIT. These features are called “Algebraic Equation Library” and “Event Library,” respectively. Users can visually arrange any number of the custom algebraic equation symbols and the custom event symbols anywhere in the editing area at any time, similar to the custom reaction step symbols;
- Mass balance equations can be automatically derived, with automatic simulation.WinBEST-KIT can automatically derive mass balance equations that represent a created reaction scheme. Because of this, users do not need to be aware of the underlying simultaneous ordinary differential equations. Users also do not need to be aware of the numerical calculation methods or of programming languages because the numerical calculations are also executed automatically;
- High-speed simulations are performed using a compiler.The numerical calculations can be performed using an external compiler (see Section 6). Although the compiler needs to be installed, the cumbersome work of compilation is performed automatically thereafter. If users want to try out WinBEST-KIT, numerical calculations can be performed by the built-in interpreter;
- A wide variety of analysis functions are available.WinBEST-KIT provides a wide variety of analysis functions in addition to regular time-course simulation, including estimation of values of kinetic parameters (using the modified Powell method [24], real-coded genetic algorithms [25], and their hybrid method [26,27]), and real-time simulation through the Virtual Lab feature. Especially, estimation of the values of kinetic parameters is a useful function. It can estimate the values of kinetic parameters from experimentally observed time-course data, even if the reaction schemes include the kinetic equations, the algebraic equations, and the events. The analysis results that can be displayed include time-course graphs, phase-plane diagrams, numerical data, and mass balance equations;
- Import from and export to SBML models.SBML is a standardized file format for exchanging models describing biochemical reaction networks, which has been adopted by about 300 applications. More than 1000 SBML models that have been published are stored in the BioModels Database [28,29,30]. Users can analyze and reuse these already published models and publish their created models. Note that some features of SBML cannot be used in WinBEST-KIT (see Section 7);
- Export to MATLAB m-files.Reaction schemes created in WinBEST-KIT can be exported as MATLAB m-files, which allows users to compare the simulation results with this well-established numerical simulation application and to convert SBML models to MATLAB m-files through WinBEST-KIT. Note that reaction schemes created using functions not supported in MATLAB, such as events in SBML, cannot be exported because the exported MATLAB m-files uses the MATLAB built-in ODE solver.
3. Considerations for Multi-Layered Metabolic Pathways
4. Implementation of New Features for Analyzing Multi-Layered Metabolic Pathways
5. Examples of New Features of WinBEST-KIT
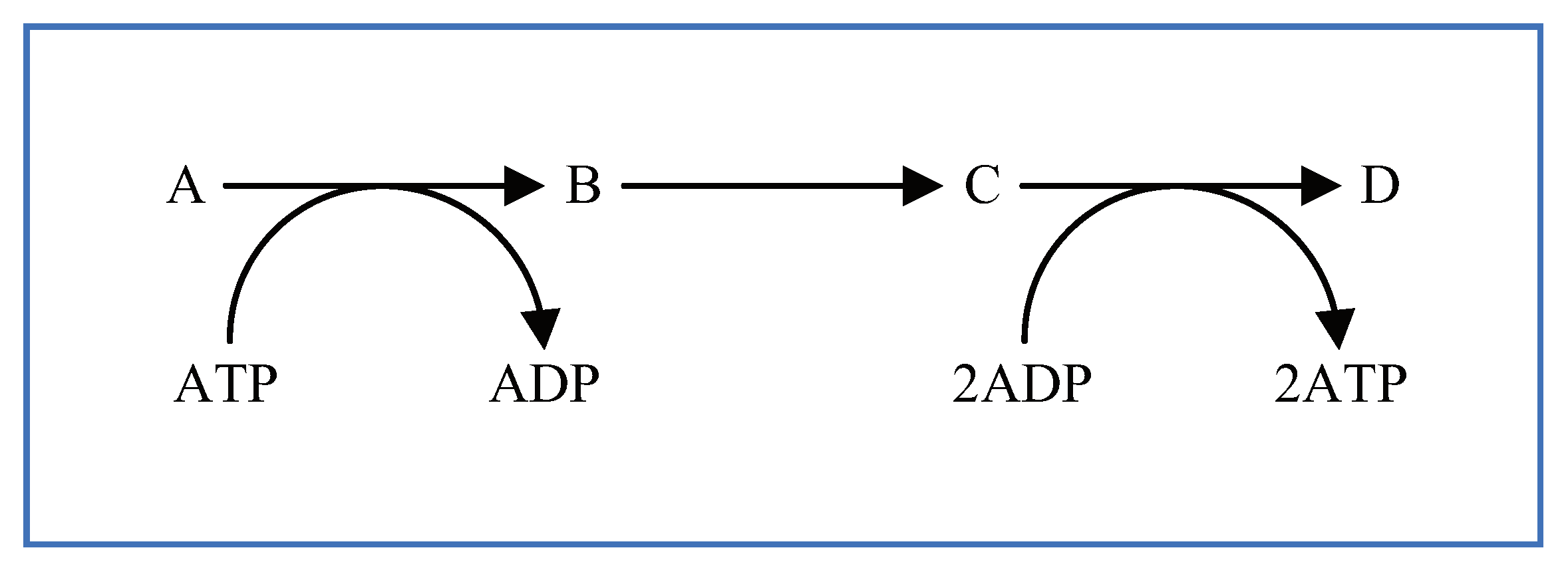
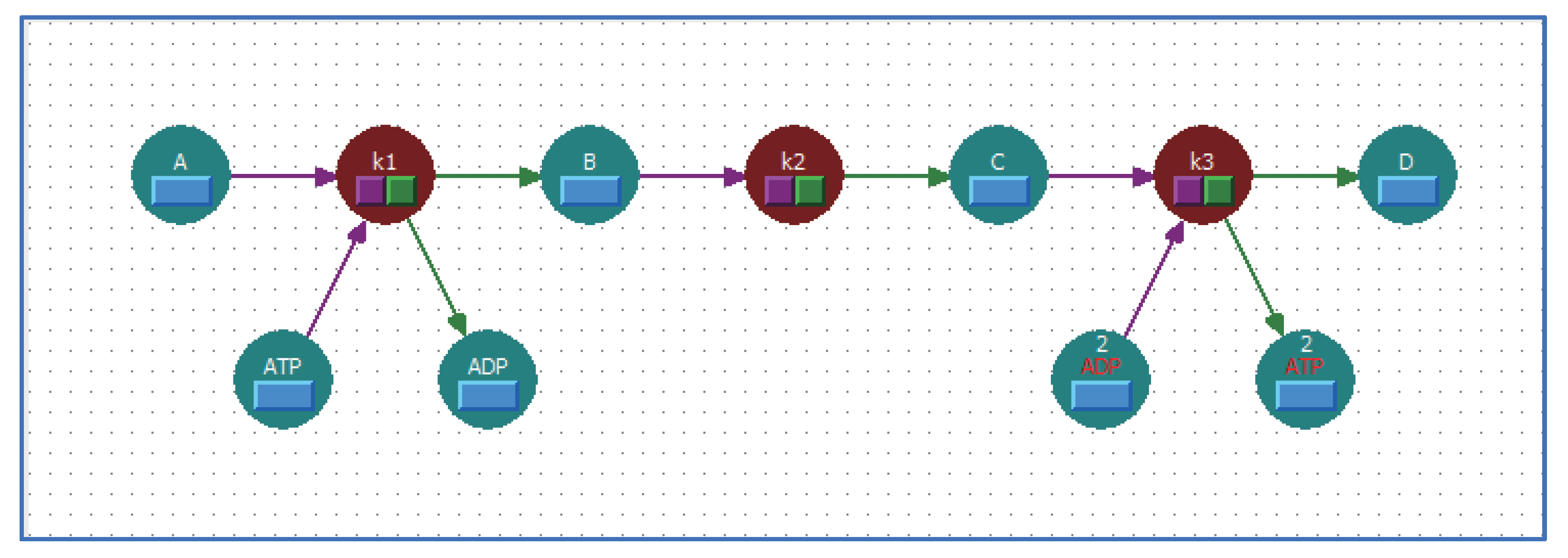
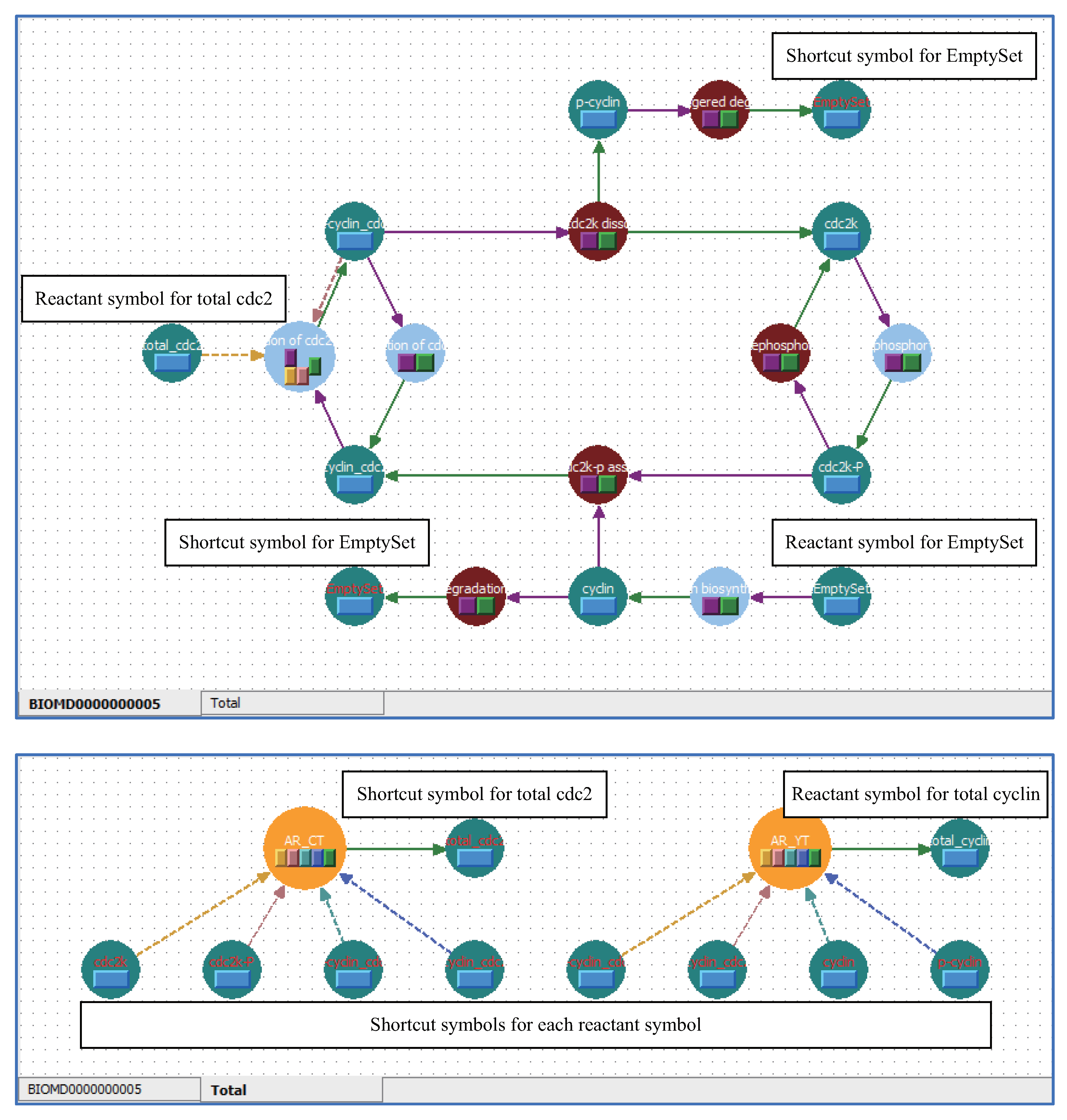
5.1. Arrange Shortcut Symbols with Unique Stoichiometric Coefficients
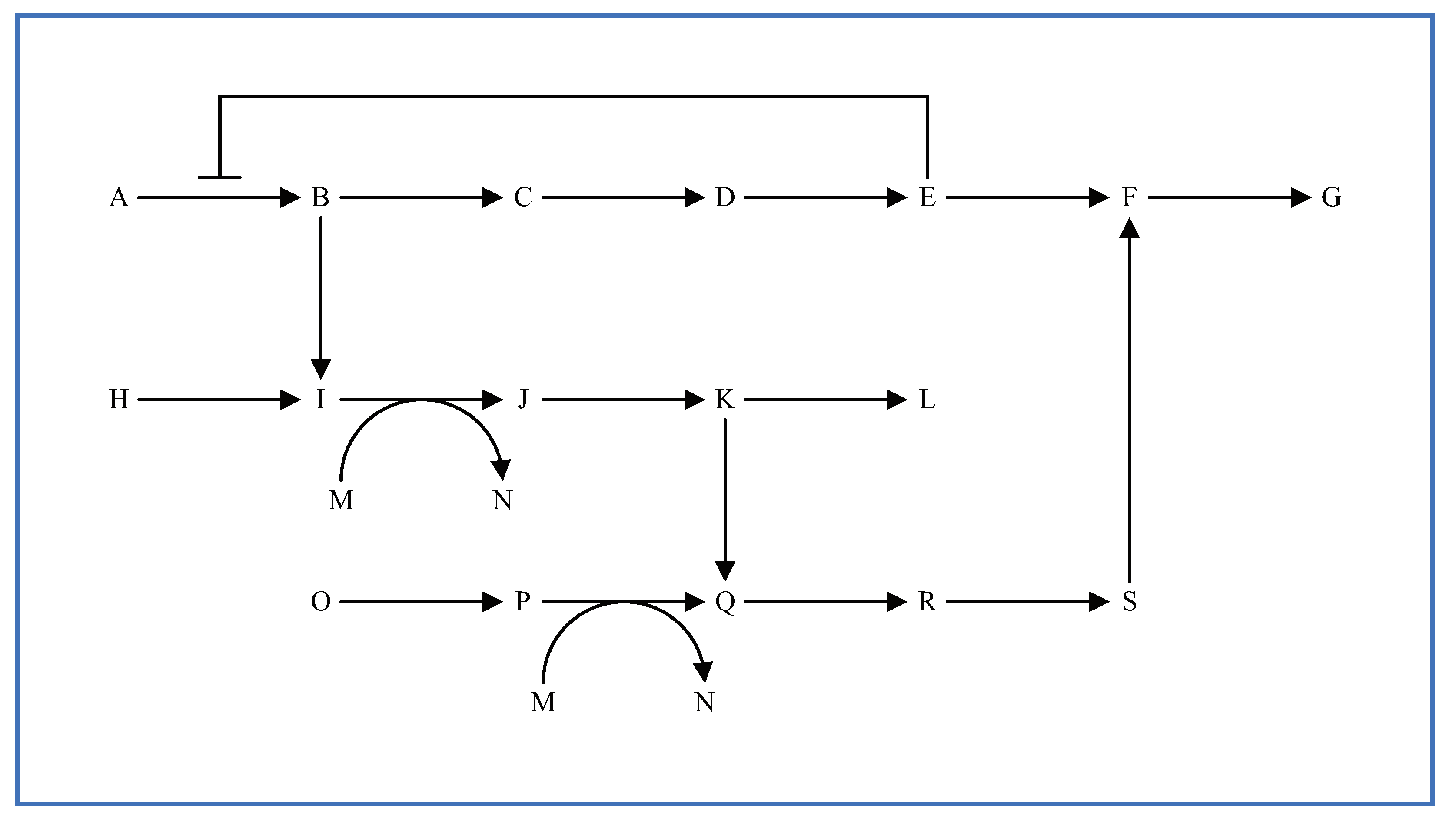
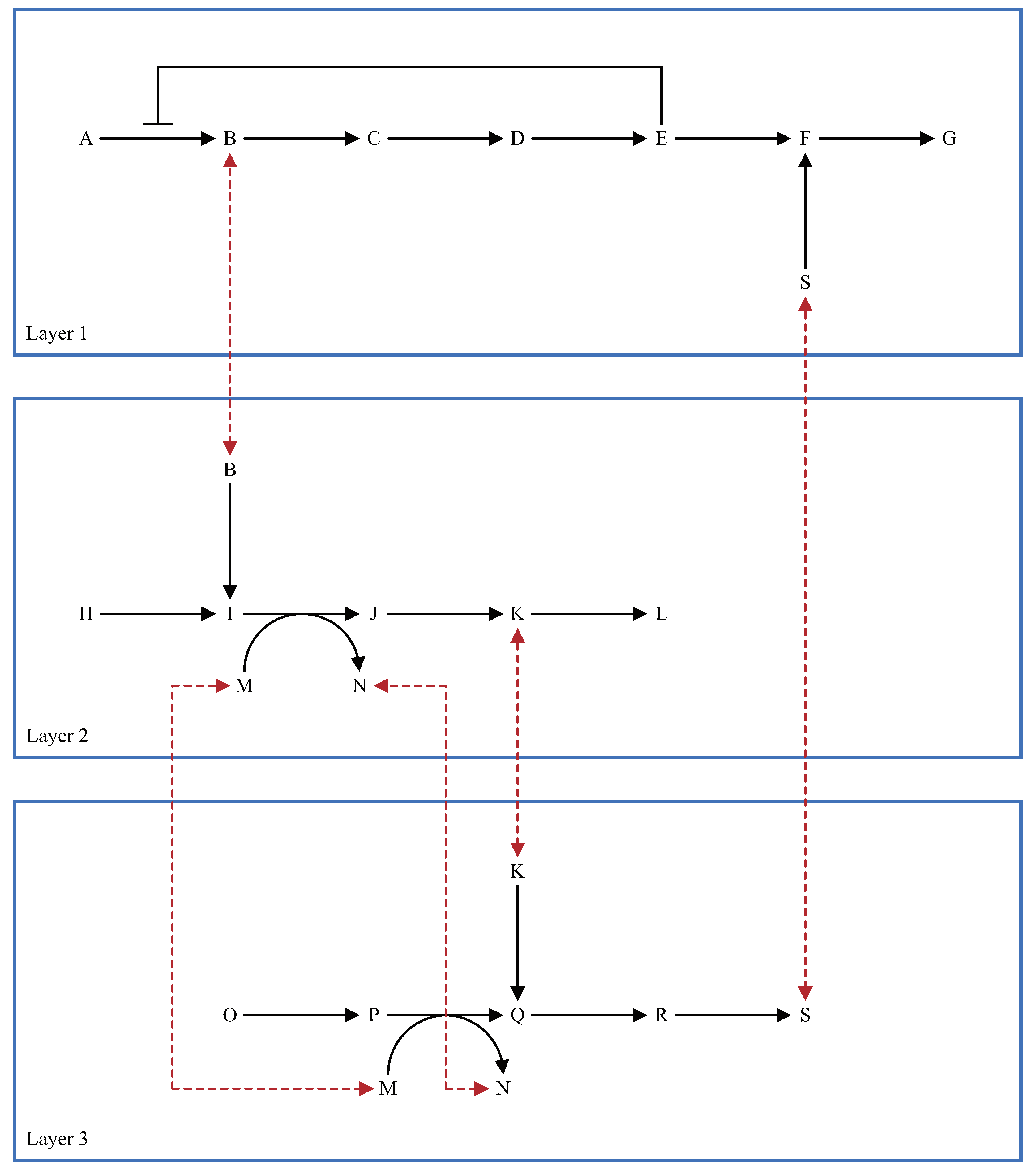
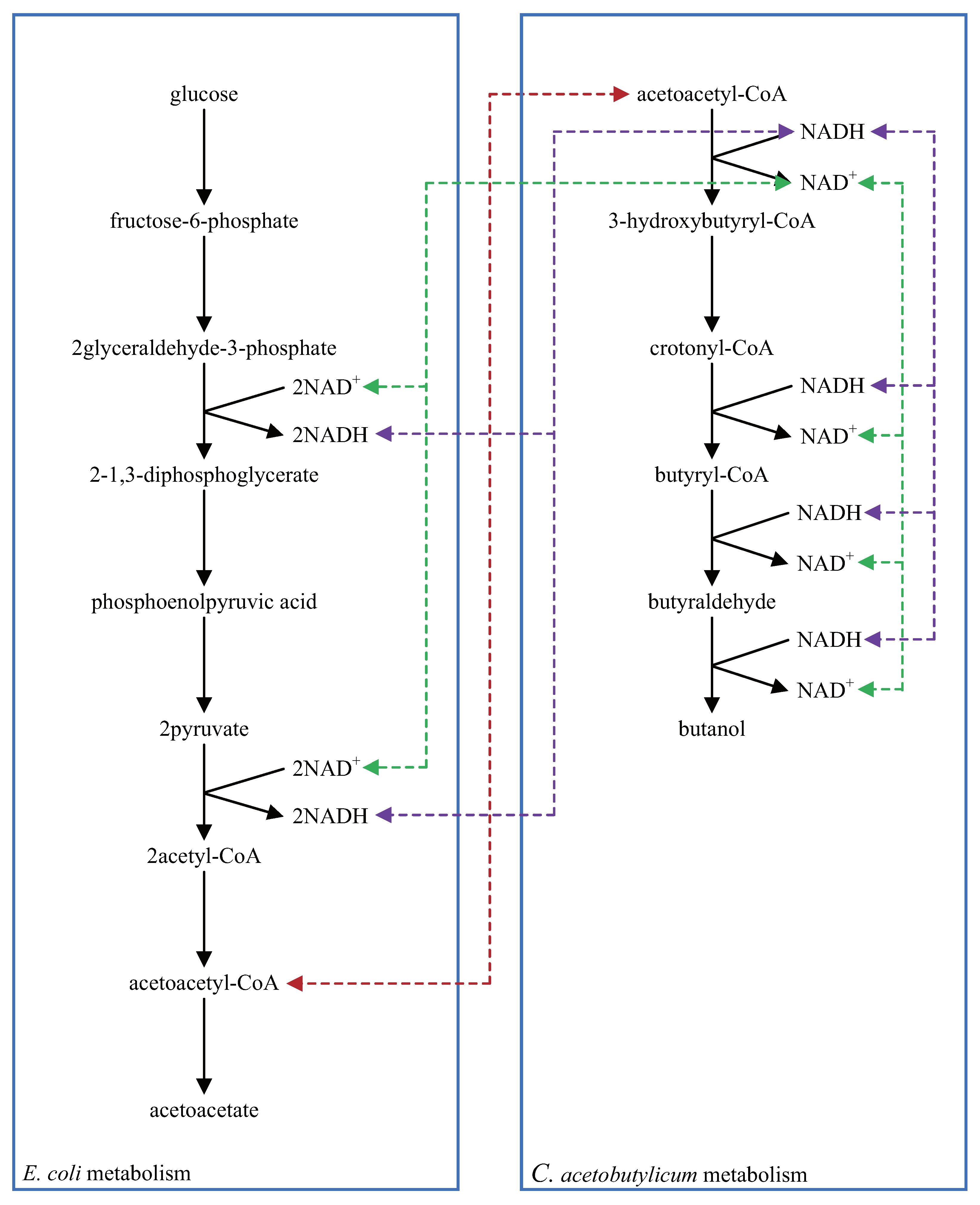
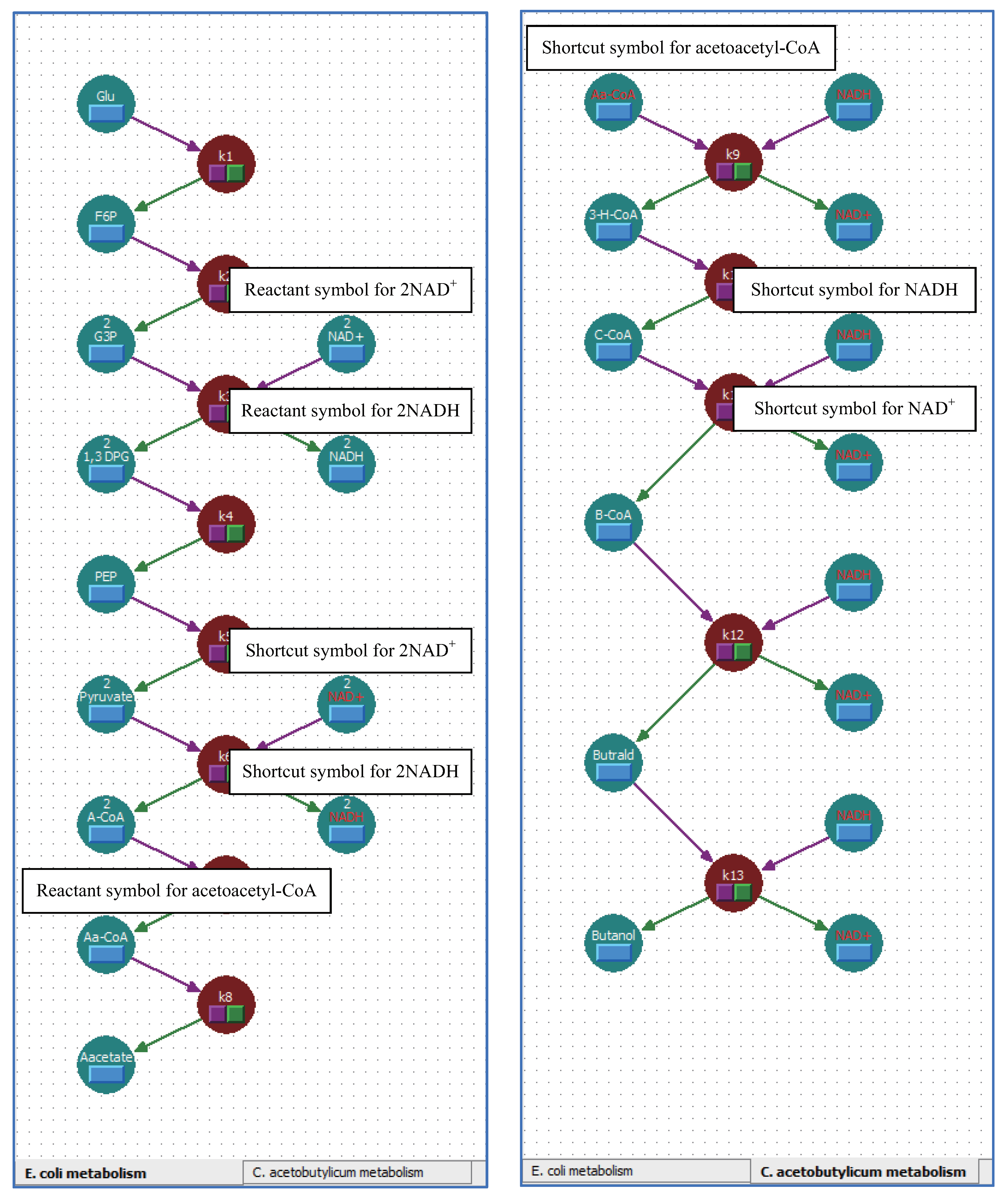
5.2. Multi-Layered Metabolic Pathways
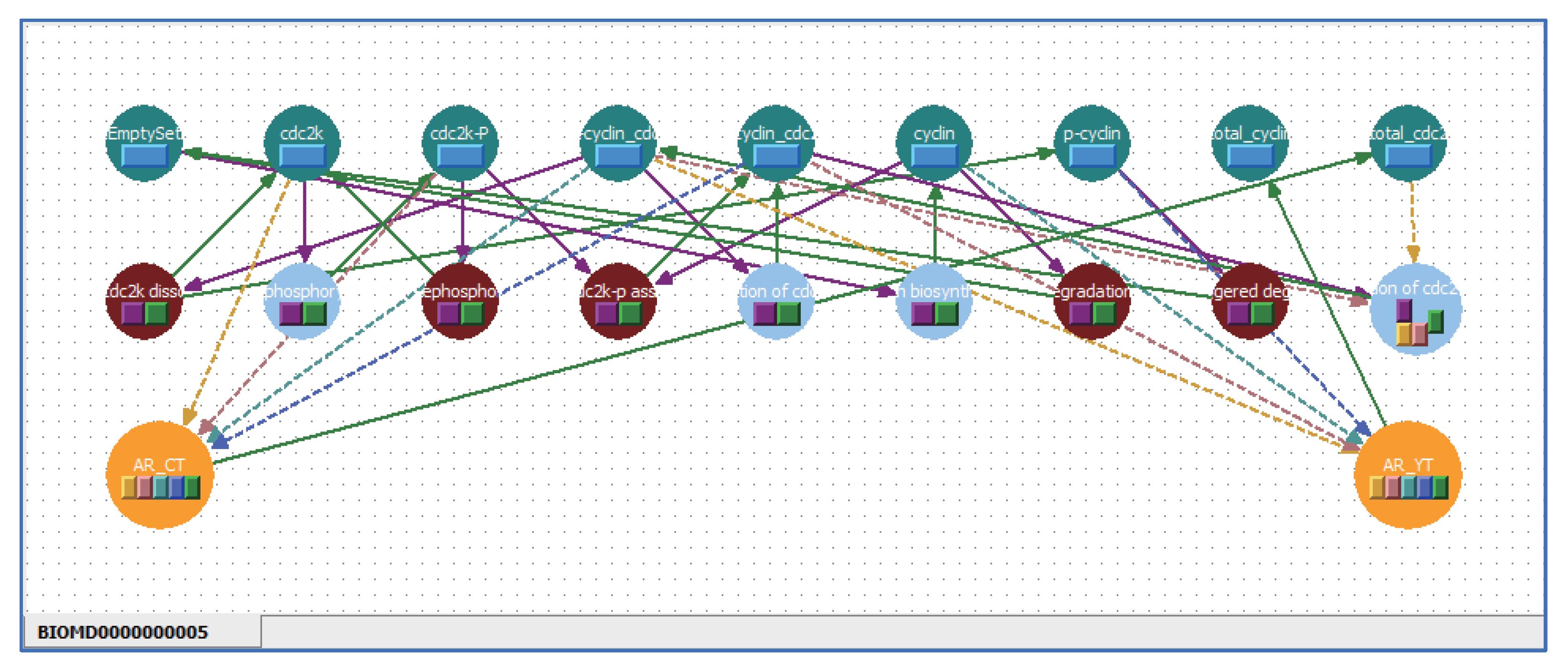
5.3. Applying Shortcut Symbols and Layers to Imported SBML Models
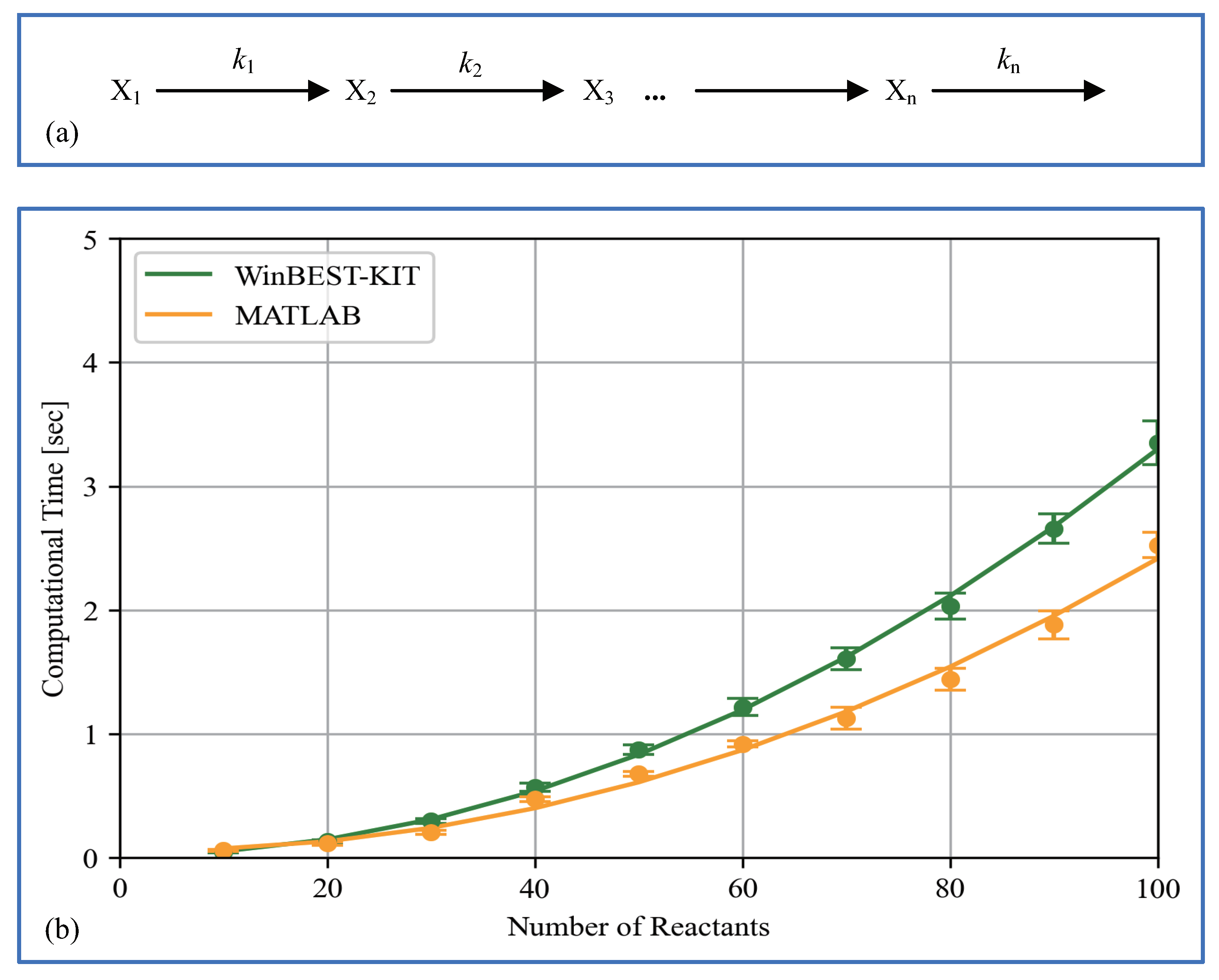
6. Performance of Computational Time
7. Future Developments
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kitano, H. Perspectives on Systems Biology. New Gener. Comput. 2000, 18, 199–216. [Google Scholar] [CrossRef]
- Kitano, H. Systems biology: A brief overview. Science 2002, 295, 1662–1664. [Google Scholar] [CrossRef] [Green Version]
- Horacio, P.S.; Afshin, F.; Jose, M.C.; Hesham, H.A.; Mario, C. Applications of High Performance Computing in Bioinformatics, Computational Biology and Computational Chemistry. In Proceedings of the 3rd International Conference on Bioinformatics and Biomedical Engineering (IWBBIO 2015), Granada, Spain, 15–17 April 2015; pp. 527–541. [Google Scholar]
- Funahashi, A.; Morohashi, M.; Kitano, H.; Tanimura, N. CellDesigner: A process diagram editor for gene-regulatory and biochemical networks. BIOSILICO 2003, 1, 159–162. [Google Scholar] [CrossRef]
- Funahashi, A.; Matsuoka, Y.; Jouraku, A.; Morohashi, M. CellDesigner 3.5: A Versatile Modeling Tool for Biochemical Networks. Proc. IEEE 2008, 96, 1254–1265. [Google Scholar] [CrossRef]
- Hoops, S.; Sahle, S.; Gauges, R.; Lee, C.; Pahle, J.; Simus, N.; Singhal, M.; Xu, L.; Mendes, P.; Kummer, U. COPASI—A COmplex PAthway SImulator. Bioinformatics 2006, 22, 3067–3074. [Google Scholar] [CrossRef] [Green Version]
- Mendes, P.; Hoops, S.; Sahle, S.; Gauges, R.; Dada, J.; Kummer, U. Computational modeling of biochemical networks using COPASI. Methods Mol. Biol. 2009, 500, 17–59. [Google Scholar] [PubMed]
- Bergmann, F.T.; Hoops, S.; Klahn, B.; Kummer, U.; Mendes, P.; Pahle, J.; Sahle, S. COPASI and its applications in biotechnology. J. Biotechnol. 2017, 261, 215–220. [Google Scholar] [CrossRef]
- Nagasaki, M.; Doi, A.; Matsuno, H.; Miyano, S. Genomic Object Net: I. A platform for modelling and simulating biopathways. Appl. Bioinform. 2003, 2, 181–184. [Google Scholar]
- Nagasaki, M.; Saito, A.; Euna, J.; Chen, L.; Kojima, K.; Ikeda, E.; Miyano, S. Cell Illustrator 4.0: A computational platform for systems biology. In Silico Biol. 2010, 10, 5–26. [Google Scholar] [CrossRef]
- Gizzatkulov, N.M.; Goryanin, I.I.; Metelkin, E.A.; Mogilevskaya, E.A.; Peskov, K.V.; Demin, O.V. DBSolve Optimum: A software package for kinetic modeling which allows dynamic visualization of simulation results. BMC Syst. Biol. 2010, 4, 109. [Google Scholar] [CrossRef] [Green Version]
- Kolpakov, F. BIOUML—Open Source Extensible Workbench for Systems Biology. In Proceedings of the 4th International Conference on Bioinformatics of Genome Regulation and Structure, Novosibirsk, Russia, 25–30 July 2004; pp. 77–80. [Google Scholar]
- Kolpakov, F.; Akberdin, I.; Kashapov, T.; Kiselev, L.; Kolmykov, S.; Kondrakhin, Y.; Kutumova, E.; Mandrik, N.; Pintus, S.; Ryabova, A.; et al. BioUML: An integrated environment for systems biology and collaborative analysis of biomedical data. Nucleic Acids Res. 2019, 47, W225–W233. [Google Scholar] [CrossRef]
- Myers, C.J.; Barker, N.; Jones, K.; Kuwahara, H.; Madsen, C.; Nguyen, N.P. iBioSim: A tool for the analysis and design of genetic circuits. Bioinformatics 2009, 25, 2848–2849. [Google Scholar] [CrossRef]
- Watanabe, L.; Nguyen, T.; Zhang, M.; Zundel, Z.; Zhang, Z.; Madsen, C.; Roehner, N.; Myers, C. iBioSim 3: A Tool for Model-Based Genetic Circuit Design. ACS Synth. Biol. 2019, 8, 1560–1563. [Google Scholar] [CrossRef]
- Ghosh, S.; Matsuoka, Y.; Asai, Y.; Hsin, K.Y.; Kitano, H. Software for systems biology: From tools to integrated platforms. Nat. Rev. Genet. 2011, 12, 821–832. [Google Scholar] [CrossRef]
- Hucka, M.; Finney, A.; Sauro, H.M.; Bolouri, H.; Doyle, J.C.; Kitano, H.; Arkin, A.P.; Bornstein, B.J.; Bray, D.; Cornish, B.A.; et al. The Systems Biology Markup Language (SBML): A Medium for Representation and Exchange of Biochemical Network Models. Bioinformatics 2003, 19, 524–531. [Google Scholar] [CrossRef]
- Sarah, M.K.; Waltemath, D.; Konig, M.; Zhang, F.; Drager, A.; Chaouiya, C.; Bergmann, F.T.; Finney, A.; Gillespie, C.S.; Helikar, T.; et al. SBML Level 3 Community members. SBML Level 3: An extensible format for the exchange and reuse of biological models. Molecluar Syst. Biol. 2020, 16, e9110. [Google Scholar]
- Sekiguchi, T.; Okamoto, M. WinBEST-KIT: Windows-based Biochemical Reaction Simulator for Metabolic Pathways. J. Bioinform. Comput. Biol. 2006, 4, 621–638. [Google Scholar] [CrossRef] [PubMed]
- Sekiguchi, T.; Hamada, H.; Okamoto, M. WinBEST-KIT: Biochemical reaction simulator that can define and customize algebraic equations and events as GUI components. J. Bioinform. Comput. Biol. 2019, 17, 1950036. [Google Scholar] [CrossRef]
- Shinto, H.; Tashiro, Y.; Yamashita, Y.; Kobayashi, G.; Sekiguchi, T.; Hanai, T.; Kuriya, Y.; Okamoto, M.; Sonomoto, K. Kinetic modeling and sensitivity analysis of acetone-butanol-ethanol production. J. Biotechnol. 2007, 131, 45–56. [Google Scholar] [CrossRef]
- Shinto, H.; Tashiro, Y.; Kobayashi, G.; Sekiguchi, T.; Hanai, T.; Kuriya, Y.; Okamoto, M.; Sonomoto, K. Kinetic modeling and sensitivity analysis of acetone-butanol-ethanol production. Process. Biochem. 2008, 43, 1452–1461. [Google Scholar] [CrossRef]
- Sakata, K.; Saito, T.; Ohyanagi, H.; Okumura, J.; Ishige, K.; Suzuki, H.; Nakamura, T.; Komatsu, S. Loss of variation of state detected in soybean metabolic and human myelomonocytic leukaemia cell transcriptional networks under external stimuli. Sci. Rep. 2016, 6, 35946. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Powell, M.J.D. An efficient method for finding the minimum of a function of several variables without calculating derivatives. Comput. J. 1964, 7, 155–162. [Google Scholar] [CrossRef]
- Komori, A.; Maki, Y.; Nakatsui, M.; Ono, I.; Okamoto, M. Efficient Numerical Optimization Algorithm Based on New Real-Coded Genetic Algorithm, AREX + JGG, and Application to the Inverse Problem in Systems Biology. Appl. Math. 2012, 3, 1463–1470. [Google Scholar] [CrossRef] [Green Version]
- Okamoto, M.; Nonaka, T.; Ochiai, S.; Tominaga, D. Nonlinear numerical optimization with use of a hybrid Genetic Algorithm incorporating the Modified Powell method. Appl. Math. Comput. 1998, 91, 63–72. [Google Scholar] [CrossRef]
- Yoshimura, J.; Shimonobou, T.; Sekiguchi, T.; Okamoto, M. Development of the parameter-fitting module for web-based biochemical reaction simulator BEST-KIT. Chem-Bio Informatics J. 2003, 3, 114–129. [Google Scholar] [CrossRef] [Green Version]
- Le Novere, N.; Bornstein, B.; Broicher, A.; Courtot, M.; Donizelli, M.; Dharuri, H.; Li, L.; Sauro, H.; Schilstra, M.; Shapiro, B.; et al. BioModels Database: A free, centralized database ofcurated, published, quantitative kinetic models of biochemical and cellular systems. Nucleic Acids Res. 2006, 34, D689–D691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, C.; Donizelli, M.; Rodriguez, N.; Dharuri, H.; Endler, L.; Chelliah, V.; Li, L.; He, E.; Henry, A.; Stefan, M.I.; et al. BioModels Database: An enhanced, curated and annotated resource for published quantitative kinetic models. BMC Syst. Biol. 2010, 4, 92. [Google Scholar] [CrossRef] [Green Version]
- Rahuman, S.; Malik, S.; Mihai, G.; Tung, V.N.N.; Krishna, T.; Matthew, G.R.; Ashley, X.; Manh, T.V.; Jinghao, M.; Matthieu, M.; et al. BioModels—15 years of sharing computational models in life science. Nucleic Acids Res. 2020, 48, D407–D415. [Google Scholar]
- Atsumi, S.; Cann, A.F.; Connor, M.R.; Shen, C.R.; Smith, K.M.; Brynildsen, M.P.; Chou, K.J.Y.; Hanai, T.; Liao, J.C. Metabolic engineering of Escherichia coli for 1-butanol production. Metab. Eng. 2008, 10, 305–311. [Google Scholar] [CrossRef] [PubMed]
- Tyson, J.J. Modeling the cell division cycle: cdc2 and cyclin interactions. Proc. Natl. Acad. Sci. USA 1991, 88, 7328–7332. [Google Scholar] [CrossRef] [Green Version]
- Gear, C.W. Numerical Initial Value Problems in Ordinary Differential Equations; Prentice-Hall: Hoboken, NJ, USA, 1971. [Google Scholar]
- Takizawa, H.; Nakamura, K.; Tabira, A.; Chikahara, Y.; Matsui, T.; Hiroi, N.; Funahashi, A. LibSBMLSim: A reference implementation of fully functional SBML simulator. Bioinformatics 2013, 29, 1474–1476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kitano, H.; Funahashi, A.; Matsuoka, Y.; Oda, K. Using process diagrams for the graphical representation of biological networks. Nat. Biotechnol. 2005, 23, 961–966. [Google Scholar] [CrossRef] [PubMed]
- Le Novere, N.; Hucka, M.; Mi, H.; Moodie, S.; Schreiber, F.; Sorokin, A.; Demir, E.; Wegner, K.; Aladjem, M.I.; Wimalaratne, S.M.; et al. The Systems Biology Graphical Notation. Nat. Biotechnol. 2009, 27, 735–741. [Google Scholar] [CrossRef] [PubMed]














Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sekiguchi, T.; Hamada, H.; Okamoto, M. WinBEST-KIT: Biochemical Reaction Simulator for Analyzing Multi-Layered Metabolic Pathways. Bioengineering 2021, 8, 114. https://doi.org/10.3390/bioengineering8080114
Sekiguchi T, Hamada H, Okamoto M. WinBEST-KIT: Biochemical Reaction Simulator for Analyzing Multi-Layered Metabolic Pathways. Bioengineering. 2021; 8(8):114. https://doi.org/10.3390/bioengineering8080114
Chicago/Turabian StyleSekiguchi, Tatsuya, Hiroyuki Hamada, and Masahiro Okamoto. 2021. "WinBEST-KIT: Biochemical Reaction Simulator for Analyzing Multi-Layered Metabolic Pathways" Bioengineering 8, no. 8: 114. https://doi.org/10.3390/bioengineering8080114