
3D-Printed Tumor-on-Chip for the Culture of Colorectal Cancer Microspheres: Mass Transport Characterization and Anti-Cancer Drug Assays


 ,
,  , and
, and 
Abstract
:
1. Introduction
2. Materials and Methods
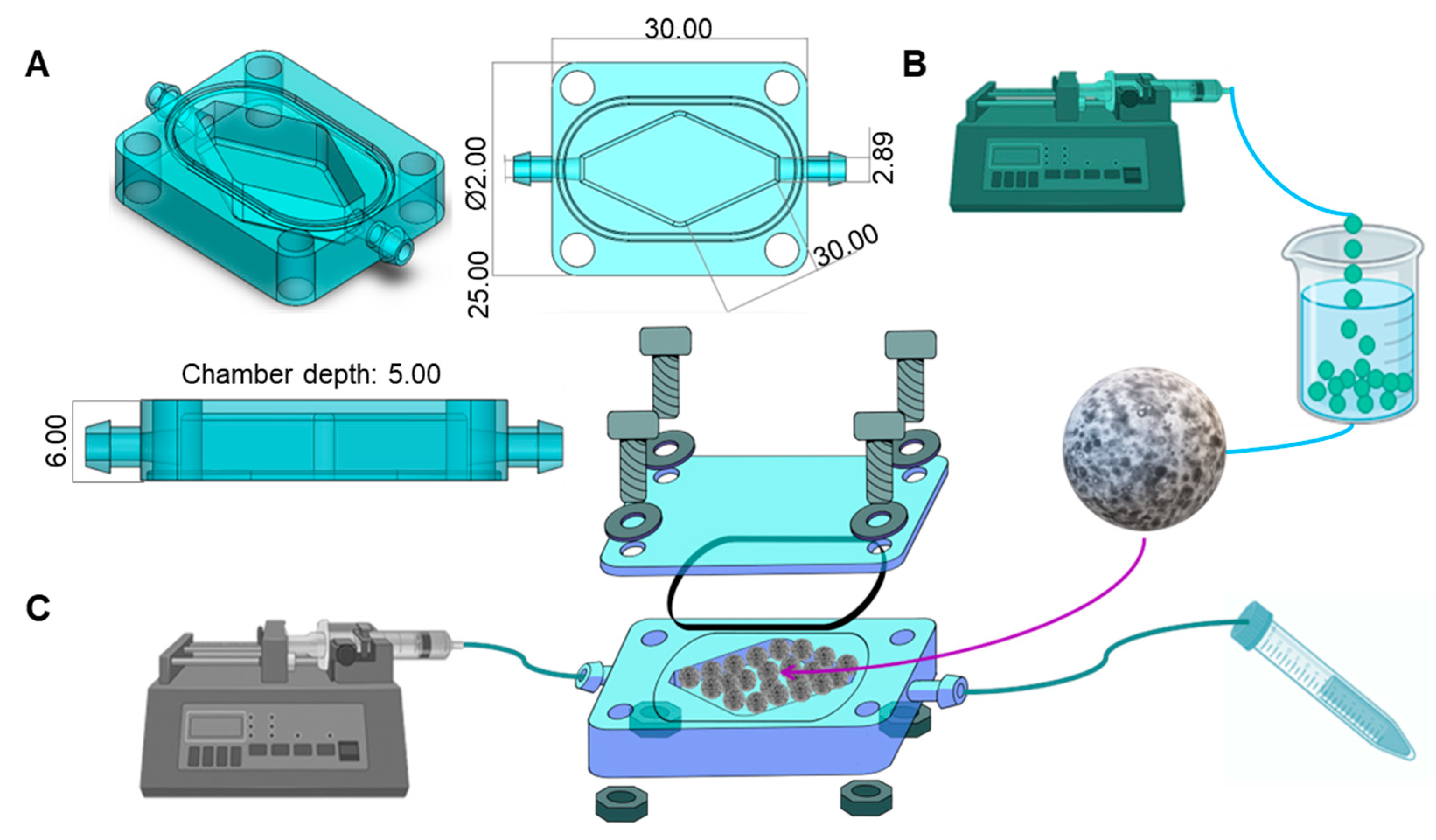
2.1. Chip Design and Fabrication
2.2. Microsphere Fabrication
2.3. FITC-Diffusion Experiment
2.4. Cell Lines and Cell Culture
2.5. Chip Assembly
2.6. Glucose Consumption Measurements
2.7. Live/Dead Analysis
2.8. In Vitro Drug Assessments
2.9. RT-qPCR
2.10. Immunostaining
2.11. Imaging and Analysis
2.12. Statistical Analysis
3. Results and Discussion
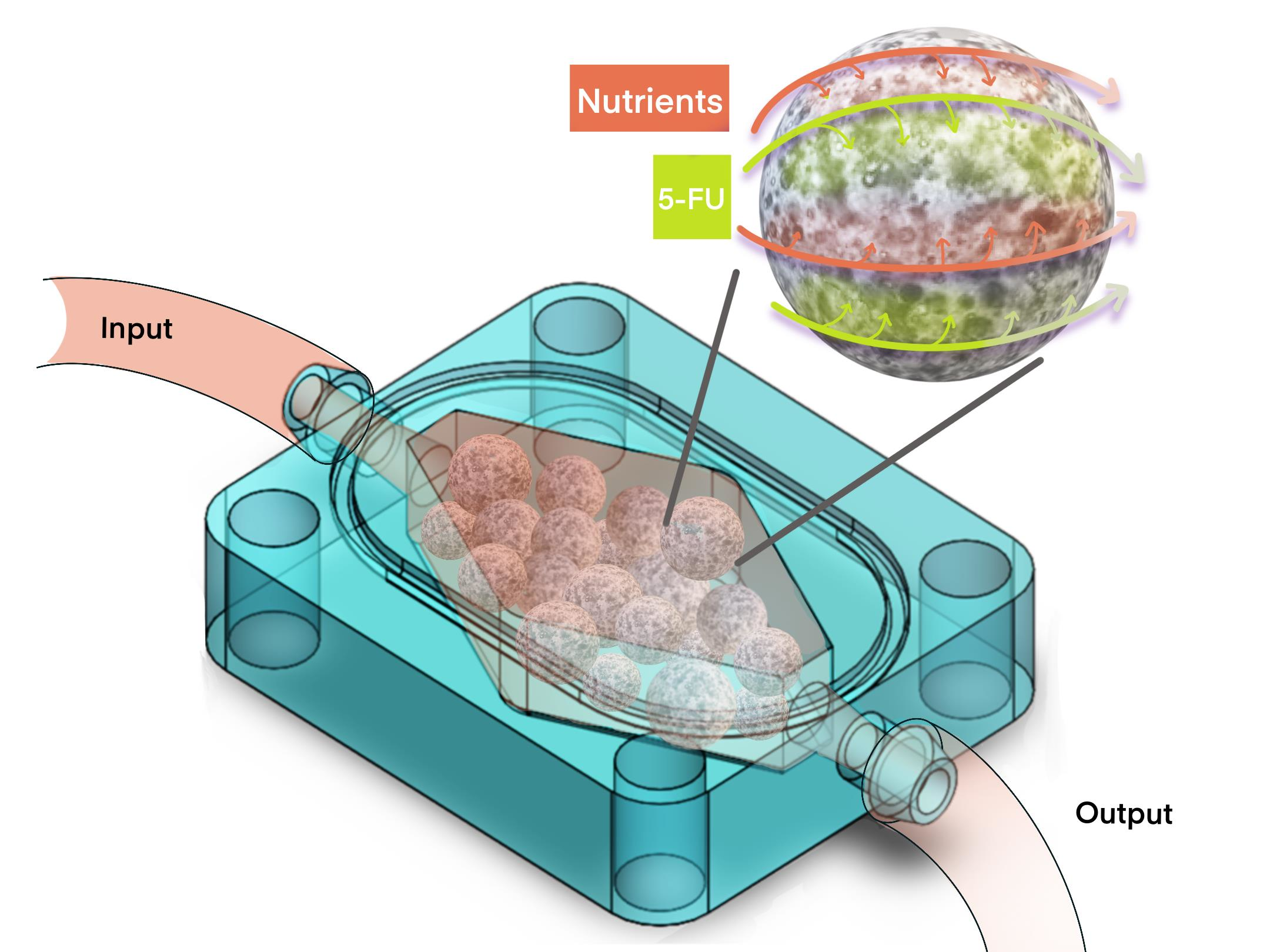
3.1. Rationale of the Design
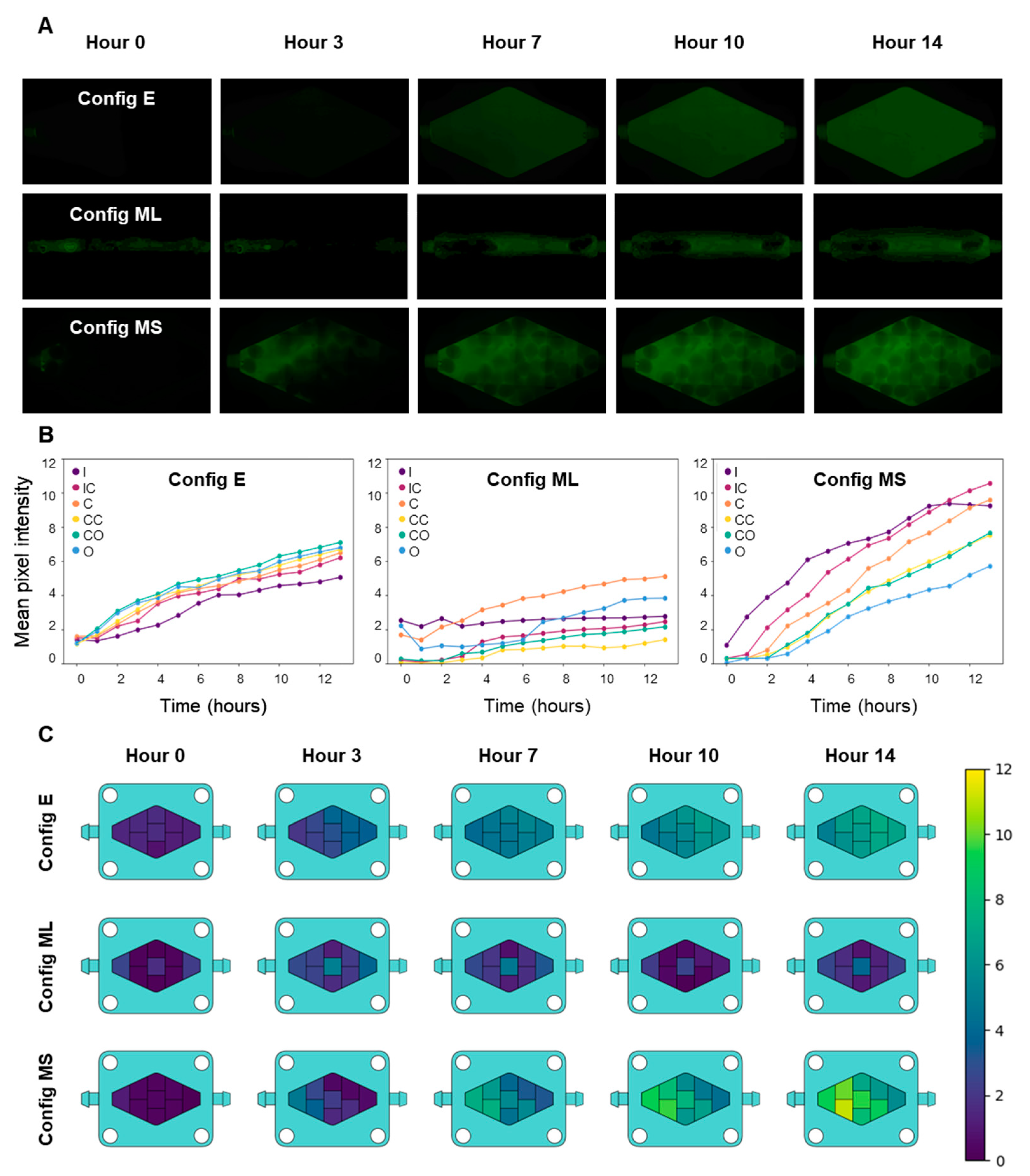
3.2. On-Chip Mixing and Transport Characterization
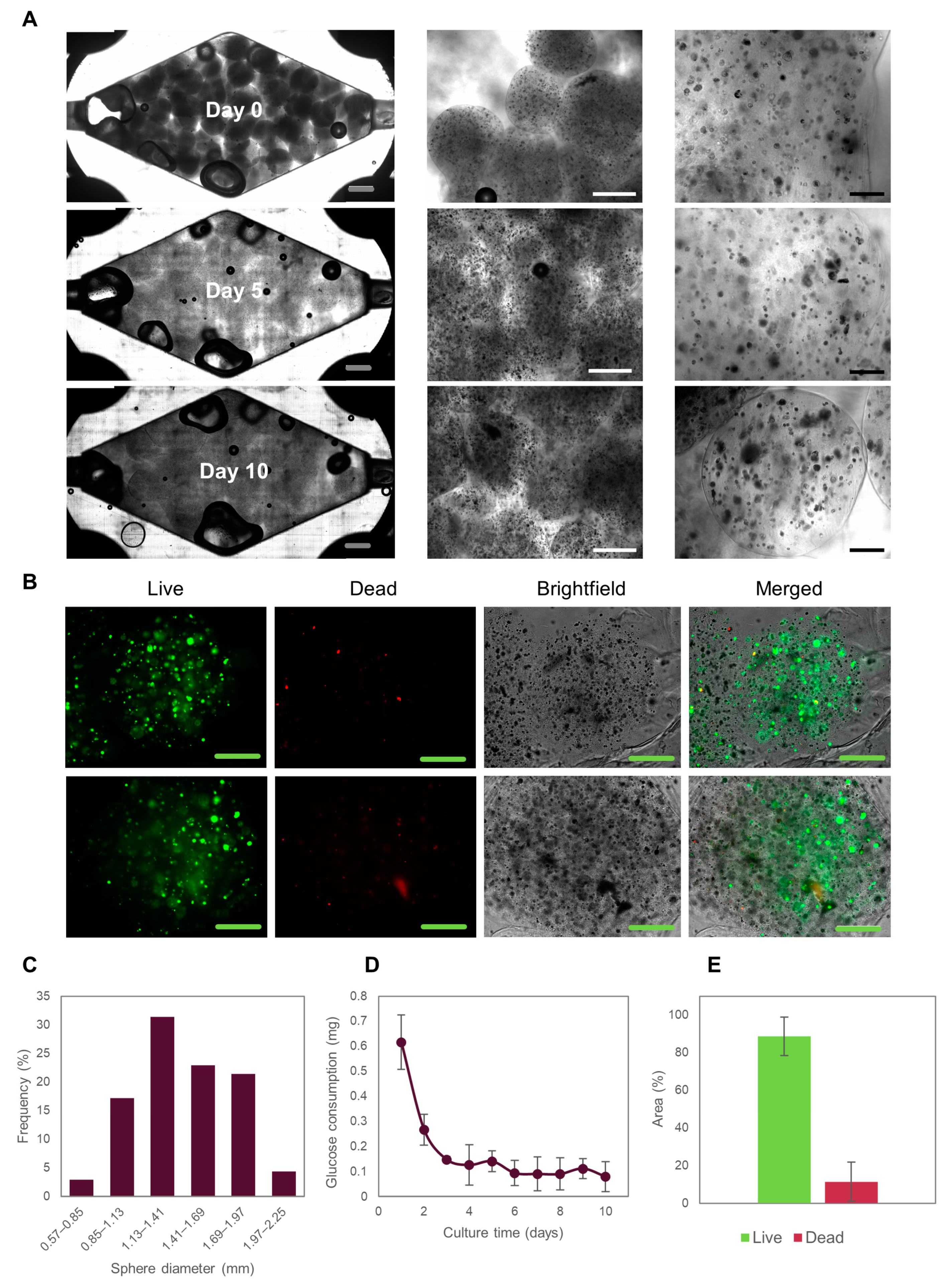
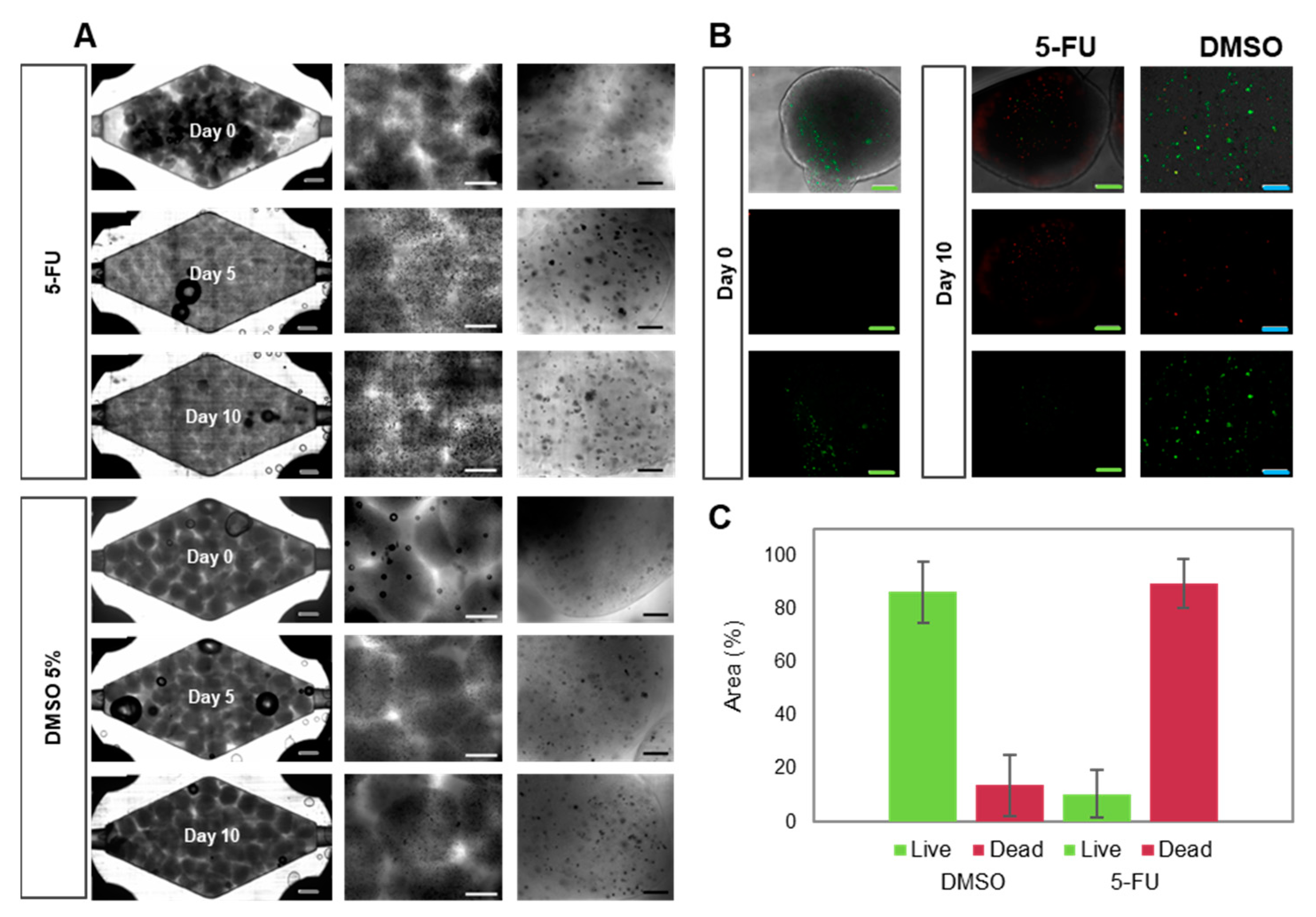
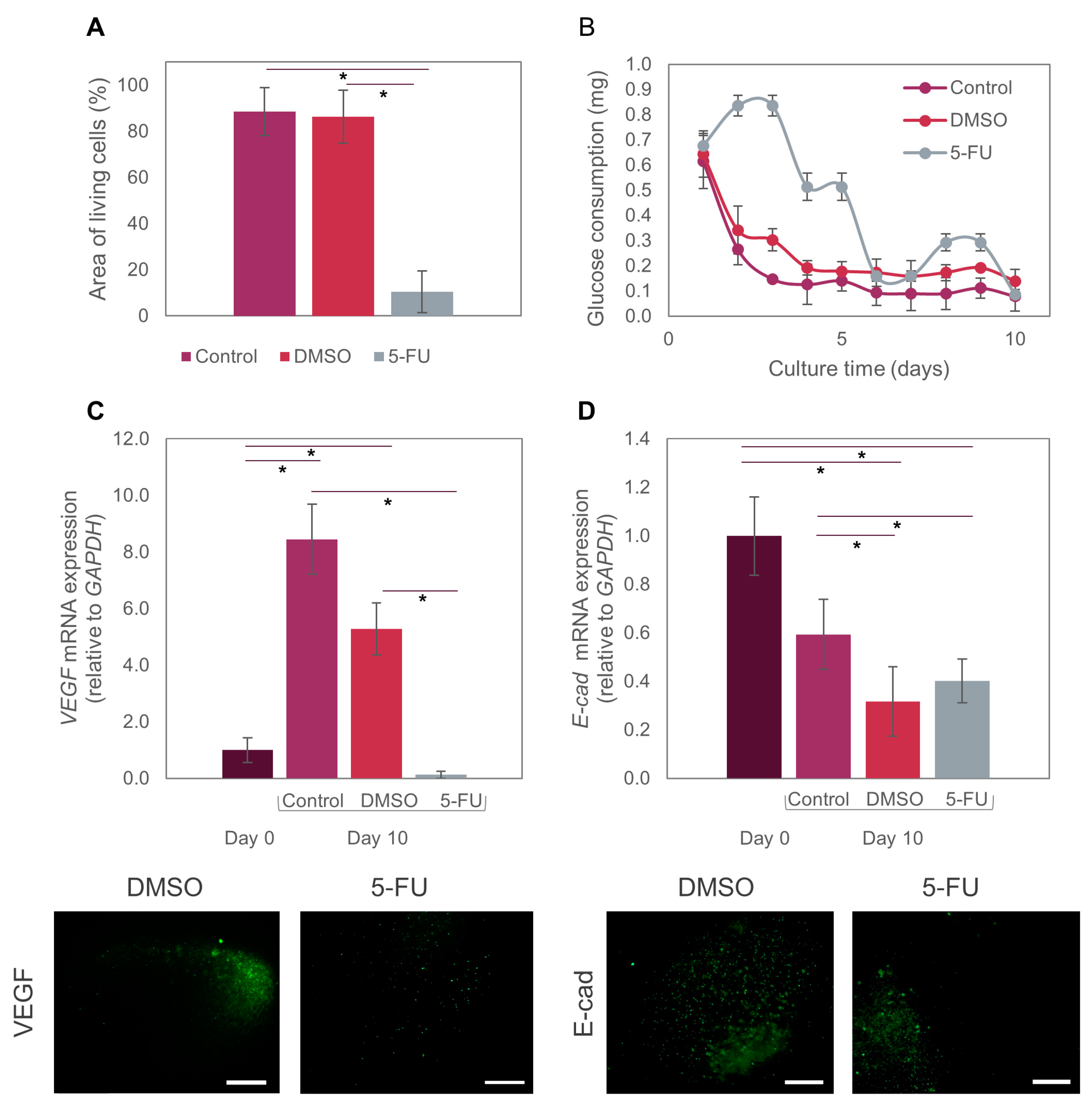
3.3. Caco2 Microtumor Survival and Development
3.4. Suitability of the Tumor-on-Chip System for Drug Testing
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ayuso, J.M.; Virumbrales-Munoz, M.; McMinn, P.H.; Rehman, S.; Gomez, I.; Karim, M.R.; Trusttchel, R.; Wisinski, K.B.; Beebe, D.J.; Skala, M.C. Tumor-on-A-chip: A microfluidic model to study cell response to environmental gradients. Lab Chip 2019, 19, 3461–3471. [Google Scholar] [CrossRef] [PubMed]
- Subia, B.; Dahiya, U.R.; Mishra, S.; Ayache, J.; Velve Casquillas, G.; Caballero, D.; Reis, R.L.; Kundu, S.C. Breast tumor-on-chip models: From disease modeling to personalized drug screening. J. Control. Release 2021, 331, 103–120. [Google Scholar] [CrossRef]
- Cekanova, M.; Rathore, K. Animal models and therapeutic molecular targets of cancer: Utility and limitations. Drug Deisgn Dev. Ther. 2014, 8, 1911–1922. [Google Scholar] [CrossRef]
- Kapałczyńska, M.; Kolenda, T.; Przybyła, W.; Zajączkowska, M.; Teresiak, A.; Filas, V.; Ibbs, M.; Bliźniak, R.; Łuczewski, Ł.; Lamperska, K. 2D and 3D cell cultures—A comparison of different types of cancer cell cultures. Arch. Med. Sci. 2016, 14, 910–919. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, M.R.; Lima, D.; Reis, R.L.; Oliveira, J.M.; Correlo, V.M. Anti-Cancer Drug Validation: The Contribution of Tissue Engineered Models. Stem Cell Rev. Rep. 2017, 13, 347–363. [Google Scholar] [CrossRef]
- Chi, C.W.; Lao, Y.H.; Ahmed, A.H.R.; Benoy, E.C.; Li, C.; Dereli-Korkut, Z.; Fu, B.M.; Leong, K.W.; Wang, S. High-Throughput Tumor-on-a-Chip Platform to Study Tumor–Stroma Interactions and Drug Pharmacokinetics. Adv. Healthc. Mater. 2020, 9, 2000880. [Google Scholar] [CrossRef]
- Liu, X.; Fang, J.; Huang, S.; Wu, X.; Xie, X.; Wang, J.; Liu, F.; Zhang, M.; Peng, Z.; Hu, N. Tumor-on-a-chip: From bioinspired design to biomedical application. Microsyst. Nanoeng. 2021, 7, 50. [Google Scholar] [CrossRef]
- Lim, J.; Ching, H.; Yoon, J.K.; Jeon, N.L.; Kim, Y.T. Microvascularized tumor organoids-on-chips: Advancing preclinical drug screening with pathophysiological relevance. Nano Converg. 2021, 8, 12. [Google Scholar] [CrossRef]
- Sobrino, A.; Phan, D.T.T.; Datta, R.; Wang, X.; Hachey, S.J.; Romero-López, M.; Gratton, E.; Lee, A.P.; George, S.C.; Hughes, C.C.W. 3D microtumors in vitro supported by perfused vascular networks. Sci. Rep. 2016, 6, 31589. [Google Scholar] [CrossRef]
- Gallegos-Martínez, S.; Lara-Mayorga, I.M.; Samandari, M.; Mendoza-Buenrostro, C.; Flores-Garza, B.G.; Reyes-Cortés, L.M.; Segoviano-Ramírez, J.C.; Zhang, Y.S.; Trujillo-de Santiago, G.; Álvarez, M.M. Culture of cancer spheroids and evaluation of anti-cancer drugs in 3D-printed miniaturized continuous stirred tank reactors (mCSTRs). Biofabrication 2022, 14, 035007. [Google Scholar] [CrossRef] [PubMed]
- Nashimoto, Y.; Okada, R.; Hanada, S.; Arima, Y.; Nishiyama, K.; Miura, T.; Yokokawa, R. Vascularized cancer on a chip: The effect of perfusion on growth and drug delivery of tumor spheroid. Biomaterials 2020, 229, 119547. [Google Scholar] [CrossRef]
- Trujillo-de-Santiago, G.; Flores-Garza, B.G.; Tavares-Negrete, J.A.; Lara-Mayorga, I.M.; González-Gamboa, I.; Zhang, Y.S.; Rojas-Martínez, A.; Ortiz-López, R.; Álvarez, M.M. The Tumor-on-Chip: Recent Advances in the Development of Microfluidic Systems to Recapitulate the Physiology of Solid Tumors. Materials 2019, 12, 2945–2985. [Google Scholar] [CrossRef] [PubMed]
- Bērziņa, S.; Harrison, A.; Taly, V.; Xiao, W. Technological advances in tumor-on-chip technology: From bench to bedside. Cancers 2021, 13, 4192. [Google Scholar] [CrossRef]
- Del Piccolo, N.; Shirure, V.S.; Bi, Y.; Goedegebuure, S.P.; Gholami, S.; Hughes, C.C.W.; Fields, R.C.; George, S.C. Tumor-on-chip modeling of organ-specific cancer and metastasis. Adv. Drug Deliv. Rev. 2021, 175, 113798. [Google Scholar] [CrossRef]
- Zhang, X.; Karim, M.; Hasan, M.M.; Hooper, J.; Wahab, R.; Roy, S.; Al-Hilal, T.A. Cancer-on-a-Chip: Models for Studying Metastasis. Cancers 2022, 14, 648. [Google Scholar] [CrossRef] [PubMed]
- Lauby-Secretan, B.; Vilahur, N.; Bianchini, F.; Guha, N.; Straif, K. The IARC Perspective on Colorectal Cancer Screening. N. Engl. J. Med. 2018, 378, 1734–1740. [Google Scholar] [CrossRef]
- Carvalho, M.R.; Barata, D.; Teixeira, L.M.; Giselbrecht, S.; Reis, R.L.; Oliveira, J.M.; Truckenmüller, R.; Habibovic, P. Colorectal tumor-on-a-chip system: A 3D tool for precision onco-nanomedicine. Sci. Adv. 2019, 5, eaaw1317. [Google Scholar] [CrossRef]
- Strelez, C.; Chilakala, S.; Ghaffarian, K.; Lau, R.; Spiller, E.; Ung, N.; Hixon, D.; Yoon, A.Y.; Sun, R.X.; Lenz, H.J.; et al. Human colorectal cancer-on-chip model to study the microenvironmental influence on early metastatic spread. iScience 2021, 24, 102509. [Google Scholar] [CrossRef]
- Habanjar, O.; Diab-Assaf, M.; Caldefie-Chezet, F.; Delort, L. 3D cell culture systems: Tumor application, advantages, and disadvantages. Int. J. Mol. Sci. 2021, 22, 12200. [Google Scholar] [CrossRef] [PubMed]
- Yue, K.; Trujillo-de Santiago, G.; Alvarez, M.M.; Tamayol, A.; Annabi, N.; Khademhosseini, A. Synthesis, properties, and biomedical applications of gelatin methacryloyl (GelMA) hydrogels. Biomaterials 2015, 73, 254–271. [Google Scholar] [CrossRef]
- Sánchez-Rodríguez, V.H. On the Improvement of the Process of Synthesis of Gelatin Methacryloyl (GelMA) Hydrogels and Development of a Hybrid Nanoparticle-GelMA-Based Bioink for Tissue Engineering. Master’s Thesis, Tecnologico de Monterrey, Monterrey, NL, Mexico, 2019. [Google Scholar]
- Bolívar-Monsalve, E.J.; Ceballos-González, C.F.; Borrayo-Montaño, K.I.; Quevedo-Moreno, D.A.; Yee-de León, J.F.; Khademhosseini, A.; Weiss, P.S.; Álvarez, M.M.; Trujillo-de Santiago, G. Continuous chaotic bioprinting of skeletal muscle-like constructs. Bioprinting 2021, 21, e00125. [Google Scholar] [CrossRef]
- Jeong, S.Y.; Lee, J.H.; Shin, Y.; Chung, S.; Kuh, H.J. Co-culture of tumor spheroids and fibroblasts in a collagen matrix-incorporated microfluidic chip mimics reciprocal activation in solid tumor microenvironment. PLoS ONE 2016, 11, e0159013. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, J.; Zhang, J.; Wu, M.; Zhang, Y. A Dynamic 3D Tumor Spheroid Chip Enables More Accurate Nanomedicine Uptake Evaluation. Adv. Sci. 2019, 6, 1901462. [Google Scholar] [CrossRef] [PubMed]
- Skardal, A.; Devarasetty, M.; Forsythe, S.; Atala, A.; Soker, S. A Reductionist Metastasis-on-a-Chip Platform for In Vitro Tumor Progression Modeling and Drug Screening. Biotechnol. Bioeng. 2016, 109, 2020–2032. [Google Scholar] [CrossRef] [PubMed]
- Sano, E.; Mori, C.; Nashimoto, Y.; Yokokawa, R.; Kotera, H.; Torisawa, Y.S. Engineering of vascularized 3D cell constructs to model cellular interactions through a vascular network. Biomicrofluidics 2018, 12, 042204. [Google Scholar] [CrossRef]
- Wan, L.; Neumann, C.; LeDuc, P. Tumor-on-a-chip for integrating a 3D tumor microenvironment: Chemical and mechanical factors. Lab Chip 2020, 20, 873–888. [Google Scholar] [CrossRef]
- Aung, A.; Kumar, V.; Theprungsirikul, J.; Davey, S.K.; Varghese, S. An Engineered Tumor-on-a-Chip Device with Breast Cancer—Immune Cell Interactions for Assessing T-cell Recruitment. Cancer Res. 2020, 80, 263–275. [Google Scholar] [CrossRef]
- Hachey, S.J.; Movsesyan, S.; Nguyen, Q.H.; Burton-Sojo, G.; Tankazyan, A.; Wu, J.; Hoang, T.; Zhao, D.; Wang, S.; Hatch, M.M.; et al. An in vitro vascularized micro-tumor model of human colorectal cancer recapitulates in vivo responses to standard-of-care therapy. Lab Chip 2021, 21, 1333–1351. [Google Scholar] [CrossRef]
- Yi, H.-G.; Jeong, Y.H.; Kim, Y.; Choi, Y.; Moon, H.E.; Park, S.H.; Kang, K.S.; Bae, M.; Jang, J.; Youn, H.; et al. A bioprinted human-glioblastoma-on-a-chip for the identification of patient-specific responses to chemoradiotherapy. Nat. Biomed. Eng. 2019, 3, 509–519. [Google Scholar] [CrossRef]
- Pedroza-González, S.C.; Rodriguez-Salvador, M.; Pérez-Benítez, B.E.; Alvarez, M.M.; Trujillo-de Santiago, G. Bioinks for 3D Bioprinting: A Scientometric Analysis of Two Decades of Progress. Int. J. Bioprint. 2021, 7, 337. [Google Scholar] [CrossRef]
- Nichol, J.W.; Koshy, S.T.; Bae, H.; Hwang, C.M.; Yamanlar, S.; Khademhosseini, A. Cell-laden microengineered gelatin methacrylate hydrogels. Biomaterials 2010, 31, 5536–5544. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Liu, J.; Zhu, W.; Tang, M.; Lawrence, N.; Yu, C.; Gou, M.; Chen, S. 3D bioprinting of functional tissue models for personalized drug screening and in vitro disease modeling. Adv. Drug Deliv. Rev. 2018, 132, 235–251. [Google Scholar] [CrossRef] [PubMed]
- Christensen, M.B.; Oberg, K.; Wolchok, J.C. Tensile properties of the rectal and sigmoid colon: A comparative analysis of human and porcine tissue. Springerplus 2015, 4, 142. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Ni, C.; Grist, S.M.; Bayly, C.; Cheung, K.C. Alginate core-shell beads for simplified three-dimensional tumor spheroid culture and drug screening. Biomed. Microdevices 2015, 17, 33. [Google Scholar] [CrossRef] [PubMed]
- Rajcevic, U.; Knol, J.C.; Piersma, S.; Bougnaud, S.; Fack, F.; Sundlisaeter, E.; Søndenaa, K.; Myklebust, R.; Pham, T.V.; Niclou, S.P.; et al. Colorectal cancer derived organotypic spheroids maintain essential tissue characteristics but adapt their metabolism in culture. Proteome Sci. 2014, 12, 39–52. [Google Scholar] [CrossRef]
- Castro, F.; Leite Pereira, C.; Helena Macedo, M.; Almeida, A.; José Silveira, M.; Dias, S.; Cardoso, A.P.; Oliveira, M.J.; Sarmento, B. Advances on colorectal cancer 3D models: The needed translational technology for nanomedicine screening. Adv. Drug Deliv. Rev. 2021, 175, 113824. [Google Scholar] [CrossRef]
- Rios De La Rosa, J.M.; Wubetu, J.; Tirelli, N.; Tirella, A. Colorectal tumor 3D in vitro models: Advantages of biofabrication for the recapitulation of early stages of tumour development. Biomed. Phys. Eng. Express 2018, 4, 045010. [Google Scholar] [CrossRef]
- Gao, J.; Zhen, R.; Liao, H.; Zhuang, W.; Guo, W. Pharmacokinetics of continuous transarterial infusion of 5-fluorouracil in patients with advanced hepatocellular carcinoma. Oncol. Lett. 2018, 15, 7175–7181. [Google Scholar] [CrossRef]
- Matsumoto, H.; Okumura, H.; Murakami, H.; Kubota, H.; Higashida, M.; Tsuruta, A.; Tohyama, K.; Hirai, T. Fluctuation in plasma 5-fluorouracil concentration during continuous 5-fluorouracil infusion for colorectal cancer. Anticancer Res. 2015, 35, 6193–6199. [Google Scholar]
- Morawska, K.; Goirand, F.; Marceau, L.; Devaux, M.; Cueff, A.; Bertaut, A.; Vincent, J.; Bengrine-Lefebre, L.; Ghiringhelli, F.; Schmitt, A. 5-FU therapeutic drug monitoring as a valuable option to reduce toxicity in patients with gastrointestinal cancer. Oncotarget 2018, 9, 11559–11571. [Google Scholar] [CrossRef]
- Abe, Y.; Sakuyama, N.; Sato, T.; Kishine, K.; Nagayasu, K.; Nakatani, A.; Kitajima, M.; Watanabe, T.; Nishimura, K.; Ochiai, T.; et al. Evaluation of the 5-fluorouracil plasma level in patients with colorectal cancer undergoing continuous infusion chemotherapy. Mol. Clin. Oncol. 2019, 11, 289–295. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Bi, Y.; Li, X.; Liu, Q.; Ma, J.; Zhang, C.; Zhang, J.; He, G. A retrospective analysis of plasma concentration monitoring of fluorouracil in patients with advanced colorectal cancer. Eur. J. Hosp. Pharm. 2020, 27, 36–40. [Google Scholar] [CrossRef] [PubMed]
- Longley, D.B.; Harkin, D.P.; Johnston, P.G. 5-Fluorouracil: Mechanisms of action and clinical strategies. Nat. Rev. Cancer 2003, 3, 330–338. [Google Scholar] [CrossRef]
- Kwapiszewska, K.; Michalczuk, A.; Rybka, M.; Kwapiszewski, R.; Brzózka, Z. A microfluidic-based platform for tumour spheroid culture, monitoring and drug screening. Lab Chip 2014, 14, 2096–2104. [Google Scholar] [CrossRef]
- Zaal, E.A.; Berkers, C.R. The influence of metabolism on drug response in cancer. Front. Oncol. 2018, 8, 500. [Google Scholar] [CrossRef] [PubMed]
- Patra, K.C.; Hay, N. The pentose phosphate pathway and cancer. Trends Biochem. Sci. 2014, 39, 347–354. [Google Scholar] [CrossRef] [PubMed]
- Bendardaf, R.; Sharif-Askari, F.S.; Sharif-Askari, N.S.; Syrjänen, K.; Pyrhönen, S. Cytoplasmic E-Cadherin Expression Is Associated with Higher Tumour Level of VEGFA, Lower Response Rate to Irinotecan-based Treatment and Poorer Prognosis in Patients with Metastatic Colorectal Cancer. Anticancer Res. 2019, 39, 1953–1957. [Google Scholar] [CrossRef] [PubMed]
- Des Guetz, G.; Uzzan, B.; Nicolas, P.; Cucherat, M.; Morere, J.F.; Benamouzig, R.; Breau, J.L.; Perret, G.Y. Microvessel density and VEGF expression are prognostic factors in colorectal cancer. Meta-analysis of the literature. Br. J. Cancer 2006, 94, 1823–1832. [Google Scholar] [CrossRef]
- Lee, S.H.; Koo, B.S.; Park, S.Y.; Kim, Y.M. Anti-angiogenic effects of resveratrol in combination with 5-fluorouracil on B16 murine melanoma cells. Mol. Med. Rep. 2015, 12, 2777–2783. [Google Scholar] [CrossRef]
- Passardi, A.; Marisi, G.; Ulivi, P. Metastatic colorectal cancer. Cancers 2021, 13, 6346. [Google Scholar] [CrossRef]
- Xu, X.; Gurski, L.A.; Zhang, C.; Harrington, D.A.; Farach-Carson, M.C.; Jia, X. Recreating the tumor microenvironment in a bilayer, hyaluronic acid hydrogel construct for the growth of prostate cancer spheroids. Biomaterials 2012, 33, 9049–9060. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Chen, Q.; Liu, W.; Zhang, J.; Wang, S.; Lin, Z.; Lin, J.M. Oxygen-induced cell migration and on-line monitoring biomarkers modulation of cervical cancers on a microfluidic system. Sci. Rep. 2015, 5, 9643. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Zhu, W.; Nowicki, M.; Miao, S.; Cui, H.; Holmes, B.; Glazer, R.I.; Zhang, L.G. 3D Bioprinting a Cell-laden Bone Matrix for Breast Cancer Metastasis Study. ACS Appl. Mater. Interfaces 2016, 8, 30017–30026. [Google Scholar] [CrossRef] [PubMed]
- Mizukami, Y.; Li, J.; Zhang, X.; Zimmer, M.A.; Iliopoulos, O.; Chung, D.C. Hypoxia-Inducible Factor-1-Independent Regulation of Vascular Endothelial Growth Factor by Hypoxia in Colon Cancer. Cancer Res. 2004, 64, 1765–1772. [Google Scholar] [CrossRef]
- Sundaramurthi, D.; Rauf, S.; Hauser, C.A.E. 3D bioprinting technology for regenerative medicine applications. Int. J. Bioprint. 2016, 2, 117–135. [Google Scholar] [CrossRef]
- Itashiki, Y.; Harada, K.; Takenawa, T.; Ferdous, T.; Ueyama, Y.; Mishima, K. Antitumor effects of bevacizumab in combination with fluoropyrimidine drugs on human oral squamous cell carcinoma. Oncol. Lett. 2021, 22, 730. [Google Scholar] [CrossRef]
- Denes, V.; Lakk, M.; Makarovskiy, A.; Jakso, P.; Szappanos, S.; Graf, L.; Mandel, L.; Karandi, I.; Geck, P. Metastasis blood test by flow cytometry: In vivo cancer spheroids and the role of hypoxia. Int. J. Cancer 2015, 136, 1528–1536. [Google Scholar] [CrossRef]
- Druzhkova, I.; Ignatova, N.; Prodanets, N.; Kiselev, N.; Zhukov, I.; Shirmanova, M.; Zagainov, V.; Zagaynova, E. E-Cadherin in Colorectal Cancer: Relation to Chemosensitivity. Clin. Color. Cancer 2019, 18, e74–e86. [Google Scholar] [CrossRef]
- Bruun, J.; Eide, P.W.; Bergsland, C.H.; Bruck, O.; Svindland, A.; Arjama, M.; Välimäki, K.; Bjørnslett, M.; Guren, M.G.; Kallioniemi, O.; et al. E-cadherin is a robust prognostic biomarker in colorectal cancer and low expression is associated with sensitivity to inhibitors of topoisomerase, aurora, and HSP90 in preclinical models. Mol. Oncol. 2022, 16, 2312–2329. [Google Scholar] [CrossRef]
- Ongaro, A.E.; Di Giuseppe, D.; Kermanizadeh, A.; Miguelez Crespo, A.; Mencattini, A.; Ghibelli, L.; Mancini, V.; Wlodarczyk, K.L.; Hand, D.P.; Martinelli, E.; et al. Polylactic is a Sustainable, Low Absorption, Low Autofluorescence Alternative to Other Plastics for Microfluidic and Organ-on-Chip Applications. Anal. Chem. 2020, 92, 6693–6701. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Forward Primer | Reverse Primer |
|---|---|---|
| VEGF-A | CACCATCGACAGAACAGTCC | GAATCCAATTCCAAGAGGGA |
| E-cadherin | CCCGCCTTATGATTCTCTGCTCGTG | TCCGTACATGTCAGCCAGCTTCTTG |
| GAPDH | GAGTCAACGGATTTGGTCG | TTGATTTTGGAGGGATCTCG |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sánchez-Salazar, M.G.; Crespo-López Oliver, R.; Ramos-Meizoso, S.; Jerezano-Flores, V.S.; Gallegos-Martínez, S.; Bolívar-Monsalve, E.J.; Ceballos-González, C.F.; Trujillo-de Santiago, G.; Álvarez, M.M. 3D-Printed Tumor-on-Chip for the Culture of Colorectal Cancer Microspheres: Mass Transport Characterization and Anti-Cancer Drug Assays. Bioengineering 2023, 10, 554. https://doi.org/10.3390/bioengineering10050554
Sánchez-Salazar MG, Crespo-López Oliver R, Ramos-Meizoso S, Jerezano-Flores VS, Gallegos-Martínez S, Bolívar-Monsalve EJ, Ceballos-González CF, Trujillo-de Santiago G, Álvarez MM. 3D-Printed Tumor-on-Chip for the Culture of Colorectal Cancer Microspheres: Mass Transport Characterization and Anti-Cancer Drug Assays. Bioengineering. 2023; 10(5):554. https://doi.org/10.3390/bioengineering10050554
Chicago/Turabian StyleSánchez-Salazar, Mónica Gabriela, Regina Crespo-López Oliver, Sofía Ramos-Meizoso, Valeri Sofía Jerezano-Flores, Salvador Gallegos-Martínez, Edna Johana Bolívar-Monsalve, Carlos Fernando Ceballos-González, Grissel Trujillo-de Santiago, and Mario Moisés Álvarez. 2023. "3D-Printed Tumor-on-Chip for the Culture of Colorectal Cancer Microspheres: Mass Transport Characterization and Anti-Cancer Drug Assays" Bioengineering 10, no. 5: 554. https://doi.org/10.3390/bioengineering10050554