
Investigation of Genome Biology by Synthetic Genome Engineering

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
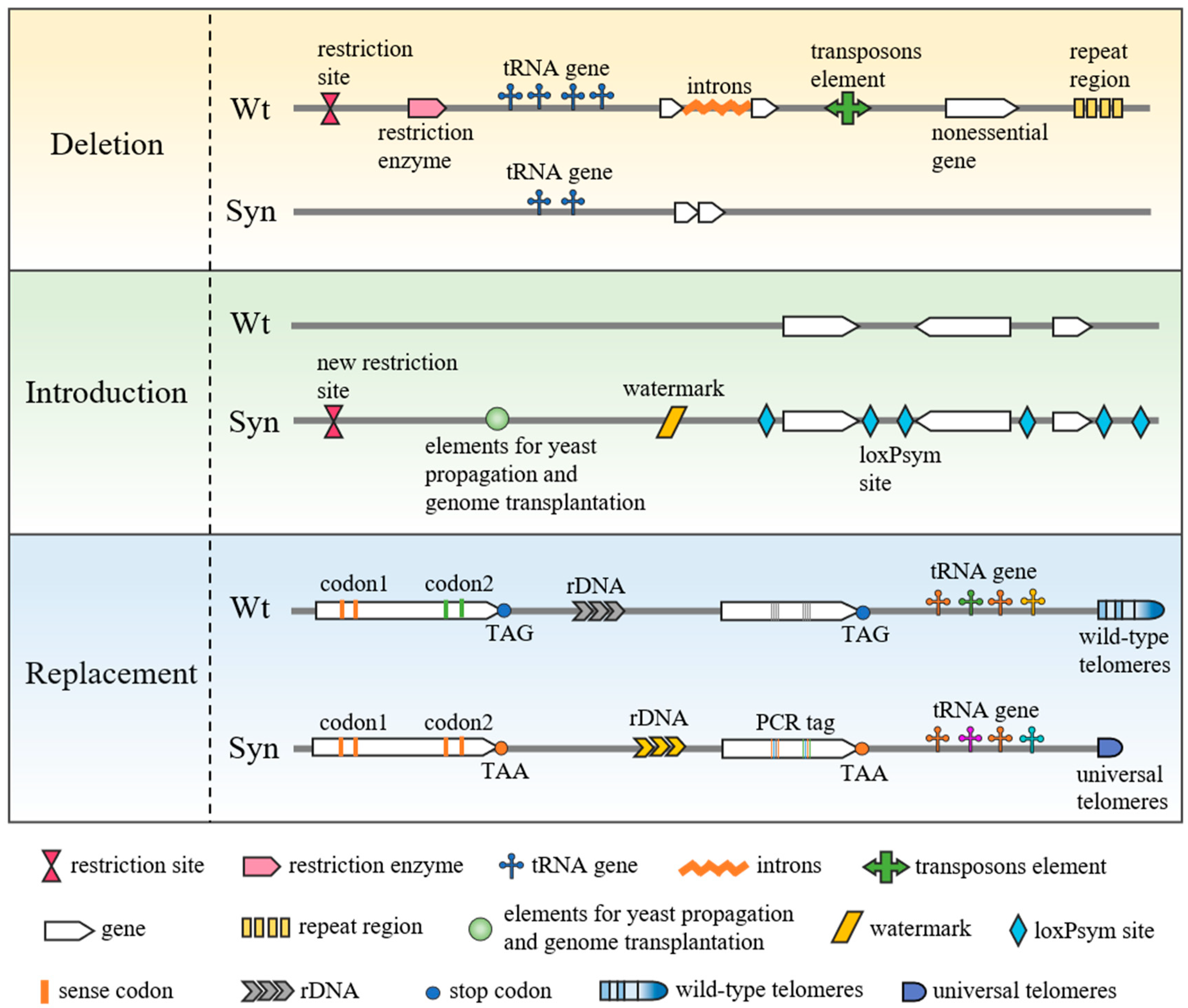
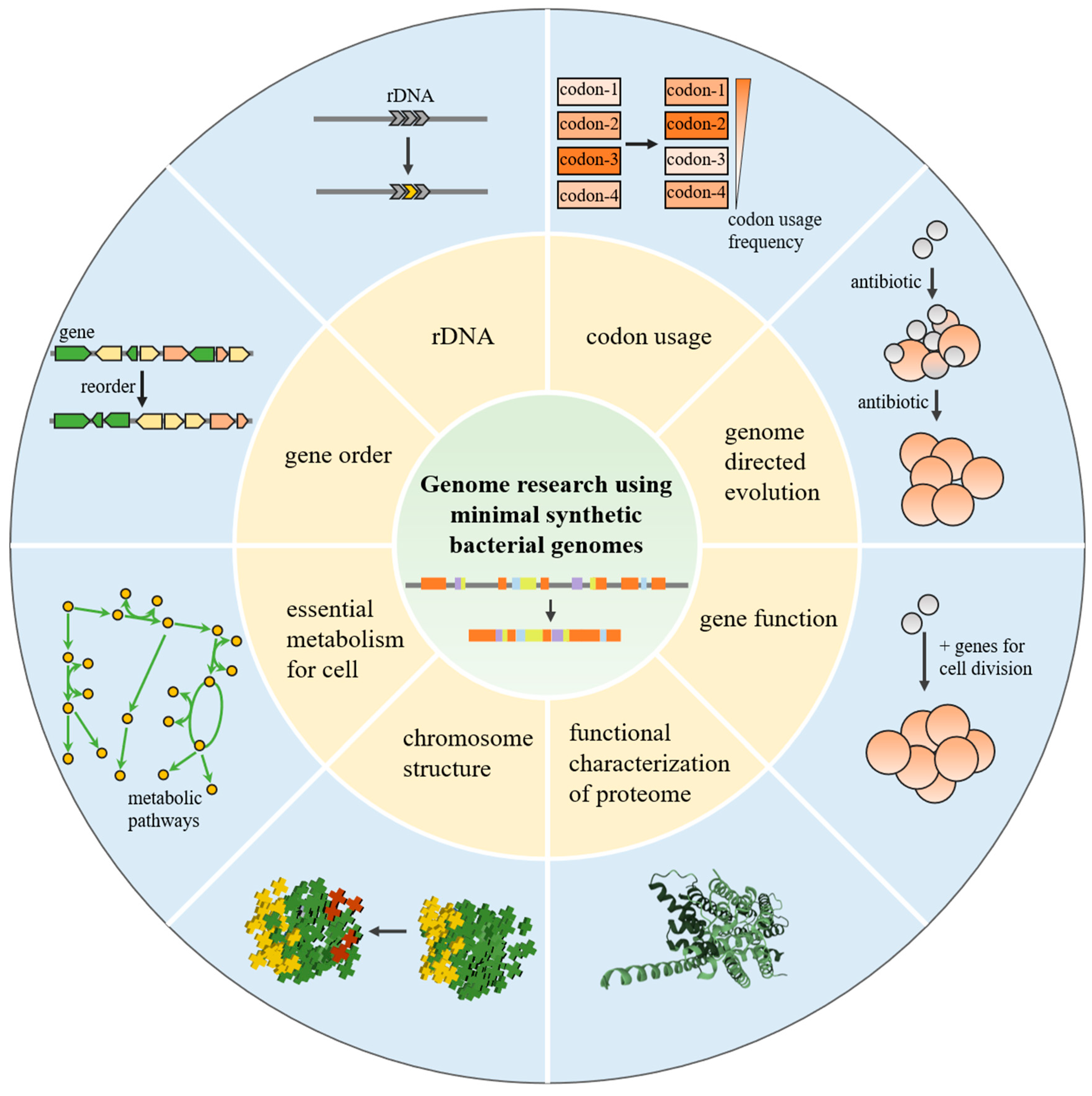
2. Genome Research Using Minimal Synthetic Bacterial Genomes
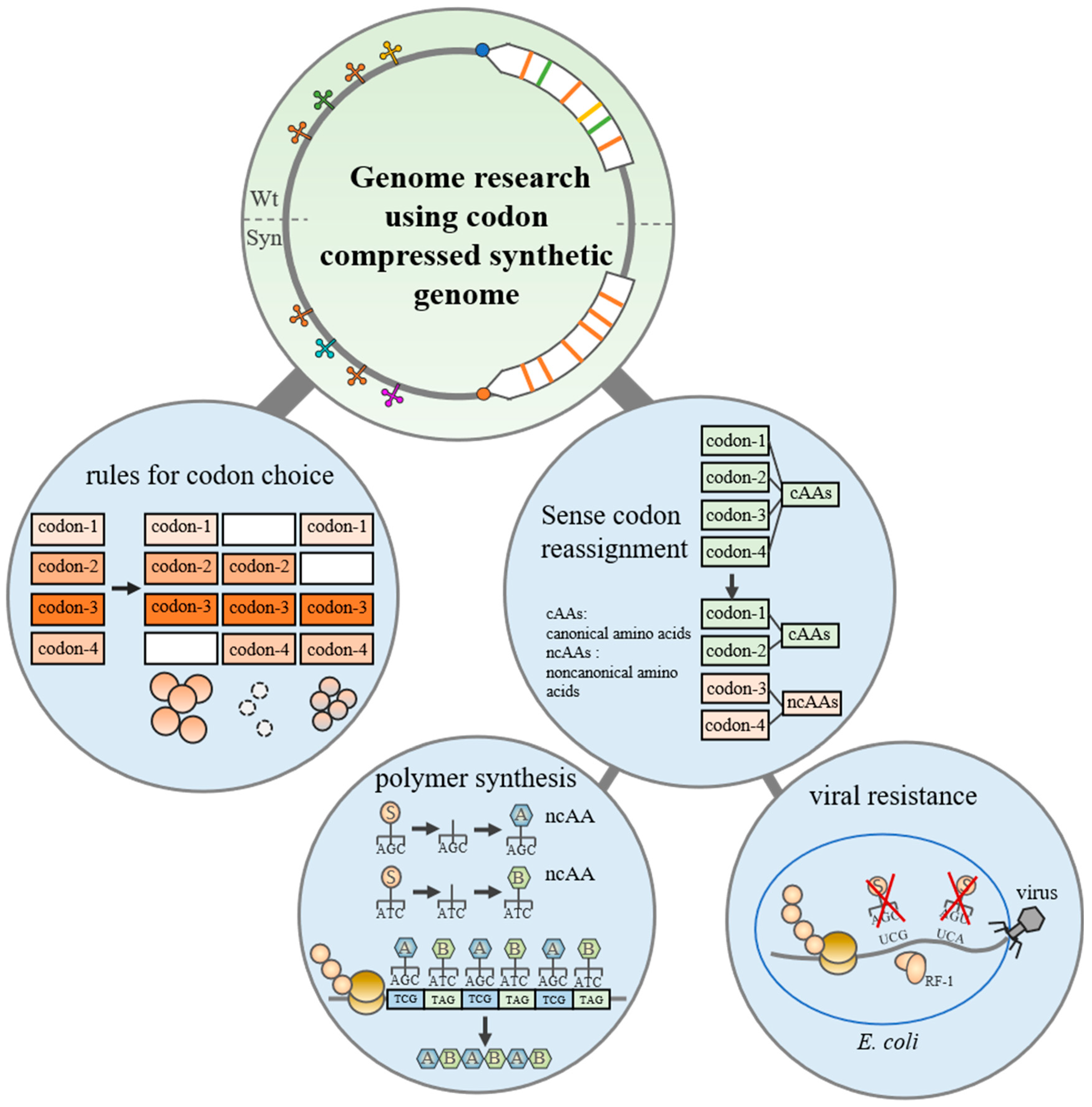
3. Genome Research Using Codon-Compressed Synthetic Genome
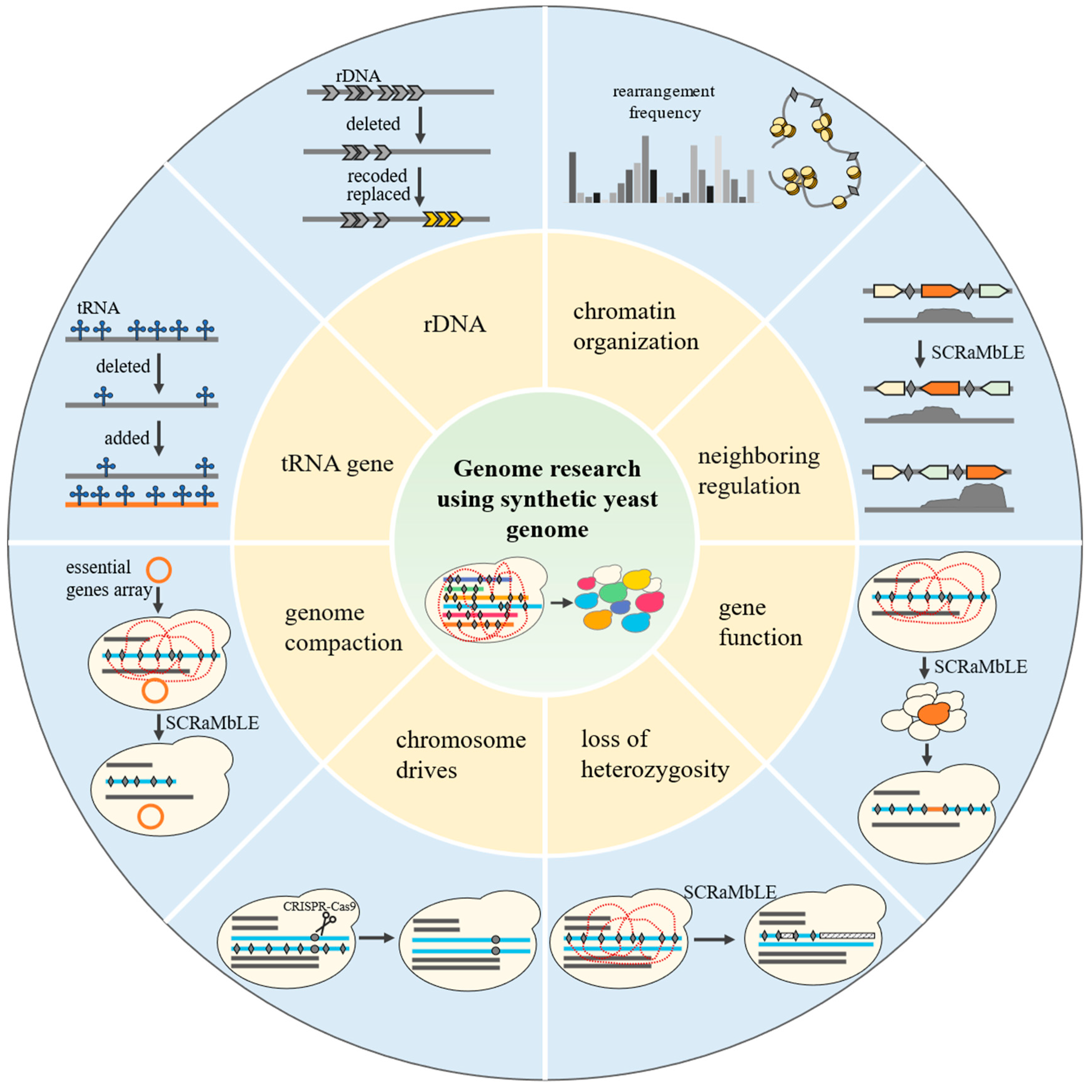
4. Genome Research Using Synthetic Yeast Genome
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Blight, J.K.; McKeating, J.A.; Marcotrigiano, J.; Rice, C.M. Efficient Replication of Hepatitis C Virus Genotype 1a RNAs in Cell Culture. J. Virol. 2003, 77, 3181–3190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blight, K.J.; Kolykhalov, A.A.; Rice, C.M. Efficient Initiation of HCV RNA Replication in Cell Culture. Science 2000, 290, 1972–1974. [Google Scholar] [CrossRef] [PubMed]
- Cello, J.; Paul, A.V.; Wimmer, E. Chemical Synthesis of Poliovirus cDNA: Generation of Infectious Virus in the Absence of Natural Template. Science 2002, 297, 1016–1018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, L.Y.; Kosuri, S.; Endy, D. Refactoring bacteriophage T7. Mol. Syst. Biol. 2005, 1, 0018. [Google Scholar] [CrossRef] [Green Version]
- Smith, H.O.; Hutchison, C.R.; Pfannkoch, C.; Venter, J.C. Generating a synthetic genome by whole genome assembly: PhiX174 bacteriophage from synthetic oligonucleotides. Proc. Natl. Acad. Sci. USA 2003, 100, 15440–15445. [Google Scholar] [CrossRef] [Green Version]
- Gibson, D.G.; Benders, G.A.; Andrews-Pfannkoch, C.; Denisova, E.A.; Baden-Tillson, H.; Zaveri, J.; Stockwell, T.B.; Brownley, A.; Thomas, D.W.; Algire, M.A.; et al. Complete chemical synthesis, assembly, and cloning of a Mycoplasma genitalium genome. Science 2008, 319, 1215–1220. [Google Scholar] [CrossRef] [Green Version]
- Gibson, D.G.; Glass, J.I.; Lartigue, C.; Noskov, V.N.; Chuang, R.Y.; Algire, M.A.; Benders, G.A.; Montague, M.G.; Ma, L.; Moodie, M.M.; et al. Creation of a bacterial cell controlled by a chemically synthesized genome. Science 2010, 329, 52–56. [Google Scholar] [CrossRef] [Green Version]
- Fredens, J.; Wang, K.; de la Torre, D.; Funke, L.F.H.; Robertson, W.E.; Christova, Y.; Chia, T.; Schmied, W.H.; Dunkelmann, D.L.; Beránek, V.; et al. Total synthesis of Escherichia coli with a recoded genome. Nature 2019, 569, 514–518. [Google Scholar] [CrossRef]
- Ostrov, N.; Landon, M.; Guell, M.; Kuznetsov, G.; Teramoto, J.; Cervantes, N.; Zhou, M.; Singh, K.; Napolitano, M.G.; Moosburner, M.; et al. Design, synthesis, and testing toward a 57-codon genome. Science 2016, 353, 819–822. [Google Scholar] [CrossRef] [Green Version]
- Shen, Y.; Gao, F.; Wang, Y.; Wang, Y.; Zheng, J.; Gong, J.; Zhang, J.; Luo, Z.; Schindler, D.; Den, Y.; et al. Dissecting Aneuploidy Phenotypes by Constructing Sc2.0 chromosome VII and SCRaMbLEing synthetic disomic yeast. bioRxiv 2022, 2, 2022-09. [Google Scholar] [CrossRef]
- Shen, Y.; Wang, Y.; Chen, T.; Gao, F.; Gong, J.; Abramczyk, D.; Walker, R.; Zhao, H.; Chen, S.; Liu, W.; et al. Deep functional analysis of synII, a 770-kilobase synthetic yeast chromosome. Science 2017, 355, 6329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Annaluru, N.; Muller, H.; Mitchell, L.A.; Ramalingam, S.; Stracquadanio, G.; Richardson, S.M.; Dymond, J.S.; Kuang, Z.; Scheifele, L.Z.; Cooper, E.M.; et al. Total Synthesis of a Functional Designer Eukaryotic Chromosome. Science 2014, 344, 55–58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, W.; Stefanita, L.L.; Yamashita, H.; Shen, M.J.; Mitchell, L.A.; Kurasawa, H.; Haase, M.A.B.; Sun, X.; Jiang, Q.; Lauer, S.L.; et al. Manipulating the 3D Organization of the Largest Synthetic Yeast Chromosome. bioRxiv 2022, 10. [Google Scholar] [CrossRef]
- Xie, Z.X.; Li, B.Z.; Mitchell, L.A.; Wu, Y.; Qi, X.; Jin, Z.; Jia, B.; Wang, X.; Zeng, B.X.; Liu, H.M.; et al. “Perfect” designer chromosome V and behavior of a ring derivative. Science 2017, 355. [Google Scholar] [CrossRef] [Green Version]
- Mitchell, L.A.; Wang, A.; Stracquadanio, G.; Kuang, Z.; Wang, X.; Yang, K.; Richardson, S.; Martin, J.A.; Zhao, Y.; Walker, R.; et al. Synthesis, debugging, and effects of synthetic chromosome consolidation: SynVI and beyond. Science 2017, 355. [Google Scholar] [CrossRef] [PubMed]
- Dymond, J.S.; Richardson, S.M.; Coombes, C.E.; Babatz, T.; Muller, H.; Annaluru, N.; Blake, W.J.; Schwerzmann, J.W.; Dai, J.; Lindstrom, D.L.; et al. Synthetic chromosome arms function in yeast and generate phenotypic diversity by design. Nature 2011, 477, 471–476. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.; Li, B.Z.; Zhao, M.; Mitchell, L.A.; Xie, Z.X.; Lin, Q.H.; Wang, X.; Xiao, W.H.; Wang, Y.; Zhou, X.; et al. Bug mapping and fitness testing of chemically synthesized chromosome X. Science 2017, 355, 1048. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, W.; Zhao, G.; Luo, Z.; Lin, Y.; Wang, L.; Guo, Y.; Wang, A.; Jiang, S.; Jiang, Q.; Gong, J.; et al. Engineering the ribosomal DNA in a megabase synthetic chromosome. Science 2017, 355, 6329. [Google Scholar] [CrossRef] [Green Version]
- Thi Nhu Thao, T.; Labroussaa, F.; Ebert, N.; V’kovski, P.; Stalder, H.; Portmann, J.; Kelly, J.; Steiner, S.; Holwerda, M.; Kratzel, A.; et al. Rapid reconstruction of SARS-CoV-2 using a synthetic genomics platform. Nature 2020, 582, 561–565. [Google Scholar] [CrossRef]
- Hekele, A.; Bertholet, S.; Archer, J.; Gibson, D.G.; Palladino, G.; Brito, L.A.; Otten, G.R.; Brazzoli, M.; Buccato, S.; Bonci, A.; et al. Rapidly produced SAM((R)) vaccine against H7N9 influenza is immunogenic in mice. Emerg. Microbes Infect. 2013, 2, e52. [Google Scholar] [CrossRef]
- Lartigue, C.; Glass, J.I.; Alperovich, N.; Pieper, R.; Parmar, P.P.; Hutchison, C.R.; Smith, H.O.; Venter, J.C. Genome transplantation in bacteria: Changing one species to another. Science 2007, 317, 632–638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hutchison, C.R.; Chuang, R.Y.; Noskov, V.N.; Assad-Garcia, N.; Deerinck, T.J.; Ellisman, M.H.; Gill, J.; Kannan, K.; Karas, B.J.; Ma, L.; et al. Design and synthesis of a minimal bacterial genome. Science 2016, 351, 6253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, S.; Feng, J.; Sun, T.; Xu, B.; Zhang, J.; Li, G.; Zhou, J.; Jiang, J. The Synthesis and Assembly of a Truncated Cyanophage Genome and Its Expression in a Heterogenous Host. Life 2022, 12, 1234. [Google Scholar] [CrossRef]
- Venetz, J.E.; Del Medico, L.; Wölfle, A.; Schächle, P.; Bucher, Y.; Appert, D.; Tschan, F.; Flores-Tinoco, C.E.; van Kooten, M.; Guennoun, R.; et al. Chemical synthesis rewriting of a bacterial genome to achieve design flexibility and biological functionality. Proc. Natl. Acad. Sci. USA 2019, 116, 8070–8079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- LeBlanc, N.; Charles, T.C. Bacterial genome reductions: Tools, applications, and challenges. Front. Genome Ed. 2022, 4. [Google Scholar] [CrossRef] [PubMed]
- Forster, A.C.; Church, G.M. Towards synthesis of a minimal cell. Mol. Syst. Biol. 2006, 2, 45. [Google Scholar] [CrossRef] [Green Version]
- Hossain, T.; Deter, H.S.; Peters, E.J.; Butzin, N.C. Antibiotic tolerance, persistence, and resistance of the evolved minimal cell, Mycoplasma mycoides JCVI-Syn3B. iScience 2021, 24, 102391. [Google Scholar] [CrossRef]
- Pelletier, J.F.; Sun, L.; Wise, K.S.; Assad-Garcia, N.; Karas, B.J.; Deerinck, T.J.; Ellisman, M.H.; Mershin, A.; Gershenfeld, N.; Chuang, R.; et al. Genetic requirements for cell division in a genomically minimal cell. Cell 2021, 184, 2430–2440. [Google Scholar] [CrossRef]
- Gilbert, B.R.; Thornburg, Z.R.; Lam, V.; Rashid, F.M.; Glass, J.I.; Villa, E.; Dame, R.T.; Luthey-Schulten, Z. Generating Chromosome Geometries in a Minimal Cell From Cryo-Electron Tomograms and Chromosome Conformation Capture Maps. Front. Mol. Biosci. 2021, 8, 644133. [Google Scholar] [CrossRef]
- Breuer, M.; Earnest, T.M.; Merryman, C.; Wise, K.S.; Sun, L.; Lynott, M.R.; Hutchison, C.A.; Smith, H.O.; Lapek, J.D.; Gonzalez, D.J.; et al. Essential metabolism for a minimal cell. Elife 2019, 8, e36842. [Google Scholar] [CrossRef]
- Bianchi, D.M.; Pelletier, J.F.; Hutchison, C.A.; Glass, J.I.; Luthey-Schulten, Z. Toward the Complete Functional Characterization of a Minimal Bacterial Proteome. J. Phys. Chem. B 2022, 126, 6820–6834. [Google Scholar] [CrossRef]
- Thornburg, Z.R.; Bianchi, D.M.; Brier, T.A.; Gilbert, B.R.; Earnest, T.M.; Melo, M.; Safronova, N.; Saenz, J.P.; Cook, A.T.; Wise, K.S.; et al. Fundamental behaviors emerge from simulations of a living minimal cell. Cell 2022, 185, 345–360. [Google Scholar] [CrossRef]
- Christen, M.; Deutsch, S.; Christen, B. Genome Calligrapher: A Web Tool for Refactoring Bacterial Genome Sequences for de Novo DNA Synthesis. Acs Synth. Biol. 2015, 4, 927–934. [Google Scholar] [CrossRef] [Green Version]
- Christen, M.; Del, M.L.; Christen, H.; Christen, B. Genome Partitioner: A web tool for multi-level partitioning of large-scale DNA constructs for synthetic biology applications. PLoS ONE 2017, 12, e177234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Kooten, M.J.F.M.; Scheidegger, C.A.; Christen, M.; Christen, B. The transcriptional landscape of a rewritten bacterial genome reveals control elements and genome design principles. Nat. Commun. 2021, 12, 3053. [Google Scholar] [CrossRef] [PubMed]
- Crick, F.H. On the genetic code. Science 1963, 139, 461–464. [Google Scholar] [CrossRef] [Green Version]
- Wang, K.; Fredens, J.; Brunner, S.F.; Kim, S.H.; Chia, T.; Chin, J.W. Defining synonymous codon compression schemes by genome recoding. Nature 2016, 539, 59–64. [Google Scholar] [CrossRef]
- Brule, C.E.; Grayhack, E.J. Synonymous Codons: Choose Wisely for Expression. Trends Genet. 2017, 33, 283–297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaney, J.L.; Clark, P.L. Roles for Synonymous Codon Usage in Protein Biogenesis. Annu. Rev. Biophys. 2015, 44, 143–166. [Google Scholar] [CrossRef] [PubMed]
- Plotkin, J.B.; Kudla, G. Synonymous but not the same: The causes and consequences of codon bias. Nat. Rev. Genet. 2011, 12, 32–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, M.; Guo, J.; Cha, J.; Chae, M.; Chen, S.; Barral, J.M.; Sachs, M.S.; Liu, Y. Non-optimal codon usage affects expression, structure and function of clock protein FRQ. Nature 2013, 495, 111–115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tuller, T.; Carmi, A.; Vestsigian, K.; Navon, S.; Dorfan, Y.; Zaborske, J.; Pan, T.; Dahan, O.; Furman, I.; Pilpel, Y. An evolutionarily conserved mechanism for controlling the efficiency of protein translation. Cell 2010, 141, 344–354. [Google Scholar] [CrossRef] [Green Version]
- Robertson, W.E.; Funke, L.F.H.; de la Torre, D.; Fredens, J.; Wang, K.; Chin, J.W. Creating custom synthetic genomes in Escherichia coli with REXER and GENESIS. Nat. Protoc. 2021, 16, 2345–2380. [Google Scholar] [CrossRef] [PubMed]
- Kuzmin, A.A.; Tomilin, A.N. Building Blocks of Artificial CRISPR-Based Systems beyond Nucleases. Int. J. Mol. Sci. 2022, 24, 397. [Google Scholar] [CrossRef] [PubMed]
- Wright, B.W.; Molloy, M.P.; Jaschke, P.R. Overlapping genes in natural and engineered genomes. Nat. Rev. Genet. 2022, 23, 154–168. [Google Scholar] [CrossRef]
- Krakauer, D.C. Stability and evolution of overlapping genes. Evolution 2000, 54, 731–739. [Google Scholar] [CrossRef] [PubMed]
- Huvet, M.; Stumpf, M.P. Overlapping genes: A window on gene evolvability. BMC Genom. 2014, 15, 721. [Google Scholar] [CrossRef] [Green Version]
- Spanjaard, R.A.; van Duin, J. Translation of the sequence AGG-AGG yields 50% ribosomal frameshift. Proc. Natl. Acad. Sci. USA 1988, 85, 7967–7971. [Google Scholar] [CrossRef] [Green Version]
- Bonekamp, F.; Andersen, H.D.; Christensen, T.; Jensen, K.F. Codon-defined ribosomal pausing in Escherichia coli detected by using the pyrE attenuator to probe the coupling between transcription and translation. Nucleic Acids Res. 1985, 13, 4113–4123. [Google Scholar] [CrossRef] [Green Version]
- Napolitano, M.G.; Landon, M.; Gregg, C.J.; Lajoie, M.J.; Govindarajan, L.; Mosberg, J.A.; Kuznetsov, G.; Goodman, D.B.; Vargas-Rodriguez, O.; Isaacs, F.J.; et al. Emergent rules for codon choice elucidated by editing rare arginine codons in Escherichia coli. Proc. Natl. Acad. Sci. USA 2016, 113, E5588–E5597. [Google Scholar] [CrossRef] [Green Version]
- Lajoie, M.J.; Kosuri, S.; Mosberg, J.A.; Gregg, C.J.; Zhang, D.; Church, G.M. Probing the Limits of Genetic Recoding in Essential Genes. Science 2013, 342, 361–363. [Google Scholar] [CrossRef] [Green Version]
- Huber, M.; Faure, G.; Laass, S.; Kolbe, E.; Seitz, K.; Wehrheim, C.; Wolf, Y.I.; Koonin, E.V.; Soppa, J. Translational coupling via termination-reinitiation in archaea and bacteria. Nat. Commun. 2019, 10, 4006. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de la Torre, D.; Chin, J.W. Reprogramming the genetic code. Nat. Rev. Genet. 2021, 22, 169–184. [Google Scholar] [CrossRef] [PubMed]
- Arranz-Gibert, P.; Patel, J.R.; Isaacs, F.J. The Role of Orthogonality in Genetic Code Expansion. Life 2019, 9, 58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arranz-Gibert, P.; Vanderschuren, K.; Isaacs, F.J. Next-generation genetic code expansion. Curr. Opin. Chem. Biol. 2018, 46, 203–211. [Google Scholar] [CrossRef] [PubMed]
- Lajoie, M.J.; Rovner, A.J.; Goodman, D.B.; Aerni, H.R.; Haimovich, A.D.; Kuznetsov, G.; Mercer, J.A.; Wang, H.H.; Carr, P.A.; Mosberg, J.A.; et al. Genomically recoded organisms expand biological functions. Science 2013, 342, 357–360. [Google Scholar] [CrossRef] [Green Version]
- Van Raad, D.; Huber, T. In Vitro Protein Synthesis in Semipermeable Artificial Cells. ACS Synth. Biol. 2021, 10, 1237–1244. [Google Scholar] [CrossRef]
- Hibi, K.; Amikura, K.; Sugiura, N.; Masuda, K.; Ohno, S.; Yokogawa, T.; Ueda, T.; Shimizu, Y. Reconstituted cell-free protein synthesis using in vitro transcribed tRNAs. Commun. Biol. 2020, 3, 350. [Google Scholar] [CrossRef]
- Seebeck, F.P.; Ricardo, A.; Szostak, J.W. Artificial lantipeptides from in vitro translations. Chem. Commun. (Camb) 2011, 47, 6141–6143. [Google Scholar] [CrossRef]
- Doerr, A.; Foschepoth, D.; Forster, A.C.; Danelon, C. In vitro synthesis of 32 translation-factor proteins from a single template reveals impaired ribosomal processivity. Sci. Rep. 2021, 11, 1898. [Google Scholar] [CrossRef]
- Mukai, T.; Hoshi, H.; Ohtake, K.; Takahashi, M.; Yamaguchi, A.; Hayashi, A.; Yokoyama, S.; Sakamoto, K. Highly reproductive Escherichia coli cells with no specific assignment to the UAG codon. Sci. Rep.-UK 2015, 5, 9699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kipper, K.; Lundius, E.G.; Curic, V.; Nikic, I.; Wiessler, M.; Lemke, E.A.; Elf, J. Application of Noncanonical Amino Acids for Protein Labeling in a Genomically Recoded Escherichia coli. ACS Synth. Biol. 2017, 6, 233–255. [Google Scholar] [CrossRef] [PubMed]
- Robertson, W.E.; Funke, L.; de la Torre, D.; Fredens, J.; Elliott, T.S.; Spinck, M.; Christova, Y.; Cervettini, D.; Boge, F.L.; Liu, K.C.; et al. Sense codon reassignment enables viral resistance and encoded polymer synthesis. Science 2021, 372, 1057–1062. [Google Scholar] [CrossRef]
- Hodgson, D.R.; Sanderson, J.M. The synthesis of peptides and proteins containing non-natural amino acids. Chem. Soc. Rev. 2004, 33, 422–430. [Google Scholar] [CrossRef]
- Link, A.J.; Mock, M.L.; Tirrell, D.A. Non-canonical amino acids in protein engineering. Curr. Opin. Biotechnol. 2003, 14, 603–609. [Google Scholar] [CrossRef]
- Richardson, S.M.; Mitchell, L.A.; Stracquadanio, G.; Yang, K.; Dymond, J.S.; DiCarlo, J.E.; Lee, D.; Huang, C.L.V.; Chandrasegaran, S.; Cai, Y.; et al. Design of a synthetic yeast genome. Science 2017, 355, 1040–1044. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Y.; Coelho, C.; Hughes, A.L.; Lazar-Stefanita, L.; Yang, S.; Brooks, A.N.; Walker, R.S.; Zhang, W.; Lauer, S.; Hernandez, C.; et al. Debugging and consolidating multiple synthetic chromosomes reveals combinatorial genetic interactions. bioRxiv 2022, 11, 2022-04. [Google Scholar] [CrossRef]
- Zhou, S.; Wu, Y.; Zhao, Y.; Zhang, Z.; Jiang, L.; Liu, L.; Zhang, Y.; Tang, J.; Yuan, Y.J. Dynamics of synthetic yeast chromosome evolution shaped by hierarchical chromatin organization. bioRxiv 2021, 11, 2021-10. [Google Scholar] [CrossRef]
- Parenteau, J.; Durand, M.; Morin, G.; Gagnon, J.; Lucier, J.F.; Wellinger, R.J.; Chabot, B.; Elela, S.A. Introns within ribosomal protein genes regulate the production and function of yeast ribosomes. Cell 2011, 147, 320–331. [Google Scholar] [CrossRef] [Green Version]
- Parenteau, J.; Durand, M.; Veronneau, S.; Lacombe, A.A.; Morin, G.; Guerin, V.; Cecez, B.; Gervais-Bird, J.; Koh, C.S.; Brunelle, D.; et al. Deletion of many yeast introns reveals a minority of genes that require splicing for function. Mol. Biol. Cell 2008, 19, 1932–1941. [Google Scholar] [CrossRef] [Green Version]
- Isaacs, F.J.; Carr, P.A.; Wang, H.H.; Lajoie, M.J.; Sterling, B.; Kraal, L.; Tolonen, A.C.; Gianoulis, T.A.; Goodman, D.B.; Reppas, N.B.; et al. Precise manipulation of chromosomes in vivo enables genome-wide codon replacement. Science 2011, 333, 348–353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dymond, J.; Boeke, J. The Saccharomyces cerevisiae SCRaMbLE system and genome minimization. Bioengineered 2012, 3, 168–171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gottschling, D.E.; Aparicio, O.M.; Billington, B.L.; Zakian, V.A. Position effect at S. cerevisiae telomeres: Reversible repression of Pol II transcription. Cell 1990, 63, 751–762. [Google Scholar] [CrossRef]
- Runge, K.W.; Zakian, V.A. Introduction of extra telomeric DNA sequences into Saccharomyces cerevisiae results in telomere elongation. Mol. Cell. Biol. 1989, 9, 1488–1497. [Google Scholar] [CrossRef]
- Schindler, D.; Walker, R.S.; Jiang, S.; Brooks, A.N.; Wang, Y.; Mueller, C.A.; Cockram, C.; Luo, Y.; Garcia, A.; Schraivogel, D.; et al. Design, Construction, and Functional Characterization of a tRNA Neochromosome in Yeast. bioRxiv 2022, 3, 2022-10. [Google Scholar] [CrossRef]
- Mercy, G.; Mozziconacci, J.; Scolari, V.F.; Yang, K.; Zhao, G.; Thierry, A.; Luo, Y.; Mitchell, L.A.; Shen, M.; Shen, Y.; et al. 3D organization of synthetic and scrambled chromosomes. Science 2017, 355. [Google Scholar] [CrossRef] [Green Version]
- Hieter, P.; Mann, C.; Snyder, M.; Davis, R.W. Mitotic stability of yeast chromosomes: A colony color assay that measures nondisjunction and chromosome loss. Cell 1985, 40, 381–392. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Mercy, G.; Vale-Silva, L.A.; Sun, X.; Agmon, N.; Zhang, W.; Yang, K.; Stracquadanio, G.; Thierry, A.S.; Ahn, J.Y.; et al. Synthetic Chromosome Fusion: Effects on Genome Structure and Function. bioRxiv 2018, 1, 2018-08. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Fu, X.; Gong, X.; Wang, Y.; Zhang, H.; Zhao, Y.; Shen, Y. Systematic dissection of key factors governing recombination outcomes by GCE-SCRaMbLE. Nat. Commun. 2022, 13, 5836. [Google Scholar] [CrossRef]
- Shen, M.J.; Wu, Y.; Yang, K.; Li, Y.; Xu, H.; Zhang, H.; Li, B.; Li, X.; Xiao, W.; Zhou, X.; et al. Heterozygous diploid and interspecies SCRaMbLEing. Nat. Commun. 2018, 9, 1934. [Google Scholar] [CrossRef] [Green Version]
- Ma, L.; Li, Y.; Chen, X.; Ding, M.; Wu, Y.; Yuan, Y. SCRaMbLE generates evolved yeasts with increased alkali tolerance. Microb. Cell. Fact. 2019, 18, 52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, Z.; Wang, L.; Wang, Y.; Zhang, W.; Guo, Y.; Shen, Y.; Jiang, L.; Wu, Q.; Zhang, C.; Cai, Y.; et al. Identifying and characterizing SCRaMbLEd synthetic yeast using ReSCuES. Nat. Commun. 2018, 9, 1910–1930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ong, J.Y.; Swidah, R.; Monti, M.; Schindler, D.; Dai, J.; Cai, Y. SCRaMbLE: A Study of Its Robustness and Challenges through Enhancement of Hygromycin B Resistance in a Semi-Synthetic Yeast. Bioengineering 2021, 8, 42. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Xie, Z.X.; Ma, Y.; Chen, X.R.; Huang, Y.Q.; He, B.; Bin, J.; Li, B.Z.; Yuan, Y.J. Ring synthetic chromosome V SCRaMbLE. Nat. Commun. 2018, 9, 3783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jia, B.; Wu, Y.; Li, B.; Mitchell, L.A.; Liu, H.; Pan, S.; Wang, J.; Zhang, H.; Jia, N.; Li, B.; et al. Precise control of SCRaMbLE in synthetic haploid and diploid yeast. Nat. Commun. 2018, 9, 1913–1933. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Y.; Zhu, R.; Mitchell, L.A.; Ma, L.; Liu, R.; Zhao, M.; Jia, B.; Xu, H.; Li, Y.; Yang, Z.; et al. In vitro DNA SCRaMbLE. Nat. Commun. 2018, 9, 1935. [Google Scholar] [CrossRef] [Green Version]
- Wightman, E.L.I.; Kroukamp, H.; Pretorius, I.S.; Paulsen, I.T.; Nevalainen, H.K.M. Rapid Colorimetric Detection of Genome Evolution in SCRaMbLEd Synthetic Saccharomyces cerevisiae Strains. Microorganisms 2020, 8, 1914. [Google Scholar] [CrossRef]
- Gowers, G.O.F.; Chee, S.M.; Bell, D.; Suckling, L.; Kern, M.; Tew, D.; McClymont, D.W.; Ellis, T. Improved betulinic acid biosynthesis using synthetic yeast chromosome recombination and semi-automated rapid LC-MS screening. Nat. Commun. 2020, 11, 868. [Google Scholar] [CrossRef] [Green Version]
- Blount, B.A.; Gowers, G.F.; Ho, J.; Ledesma-Amaro, R.; Jovicevic, D.; McKiernan, R.M.; Xie, Z.X.; Li, B.Z.; Yuan, Y.J.; Ellis, T. Rapid host strain improvement by in vivo rearrangement of a synthetic yeast chromosome. Nat. Commun. 2018, 9, 1932. [Google Scholar] [CrossRef]
- Liu, W.; Luo, Z.; Wang, Y.; Pham, N.T.; Tuck, L.; Perez-Pi, I.; Liu, L.; Shen, Y.; French, C.; Auer, M.; et al. Rapid pathway prototyping and engineering using in vitro and in vivo synthetic genome SCRaMbLE-in methods. Nat. Commun. 2018, 9, 1936. [Google Scholar] [CrossRef] [Green Version]
- De, S.; Babu, M.M. Genomic neighbourhood and the regulation of gene expression. Curr. Opin. Cell Biol. 2010, 22, 326–333. [Google Scholar] [CrossRef] [PubMed]
- Brooks, A.N.; Hughes, A.L.; Clauder-Munster, S.; Mitchell, L.A.; Boeke, J.D.; Steinmetz, L.M. Transcriptional neighborhoods regulate transcript isoform lengths and expression levels. Science 2022, 375, 1000–1005. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.; Li, J.; Guo, Z.; Zhou, S.; Su, S.; Xiao, W.; Wu, Y.; Yuan, Y. Enhancement and mapping of tolerance to salt stress and 5-fluorocytosine in synthetic yeast strains via SCRaMbLE. Synth. Syst. Biotechnol. 2022, 7, 869–877. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wu, Y.; Ma, L.; Guo, Z.; Xiao, W.; Yuan, Y. Loss of heterozygosity by SCRaMbLEing. Sci. China Life Sci. 2019, 62, 381–393. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Han, M.; Zhou, S.; Li, B.Z.; Wu, Y.; Yuan, Y.J. Chromosome drives via CRISPR-Cas9 in yeast. Nat. Commun. 2020, 11, 4344. [Google Scholar] [CrossRef]
- Wang, P.; Xu, H.; Li, H.; Chen, H.; Zhou, S.; Tian, F.; Li, B.Z.; Bo, X.; Wu, Y.; Yuan, Y.J. SCRaMbLEing of a Synthetic Yeast Chromosome with Clustered Essential Genes Reveals Synthetic Lethal Interactions. ACS Synth. Biol. 2020, 9, 1181–1189. [Google Scholar] [CrossRef]
- Luo, Z.; Yu, K.; Xie, S.; Monti, M.; Schindler, D.; Fang, Y.; Zhao, S.; Liang, Z.; Jiang, S.; Luan, M.; et al. Compacting a synthetic yeast chromosome arm. Genome Biol. 2021, 22, 5. [Google Scholar] [CrossRef]
- Kutyna, D.R.; Onetto, C.A.; Williams, T.C.; Goold, H.D.; Paulsen, I.T.; Pretorius, I.S.; Johnson, D.L.; Borneman, A.R. Construction of a synthetic Saccharomyces cerevisiae pan-genome neo-chromosome. Nat. Commun. 2022, 13, 3628. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, H.; Xiong, Y.; Xiao, W.; Wu, Y. Investigation of Genome Biology by Synthetic Genome Engineering. Bioengineering 2023, 10, 271. https://doi.org/10.3390/bioengineering10020271
Zhang H, Xiong Y, Xiao W, Wu Y. Investigation of Genome Biology by Synthetic Genome Engineering. Bioengineering. 2023; 10(2):271. https://doi.org/10.3390/bioengineering10020271
Chicago/Turabian StyleZhang, Hui, Yao Xiong, Wenhai Xiao, and Yi Wu. 2023. "Investigation of Genome Biology by Synthetic Genome Engineering" Bioengineering 10, no. 2: 271. https://doi.org/10.3390/bioengineering10020271