
Identification of a Novel Wnt Antagonist Based Therapeutic and Diagnostic Target for Alzheimer’s Disease Using a Stem Cell-Derived Model
 ,
,
Abstract
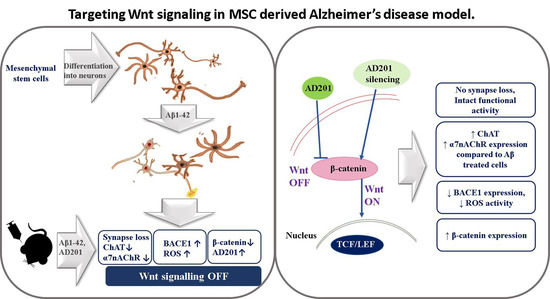
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
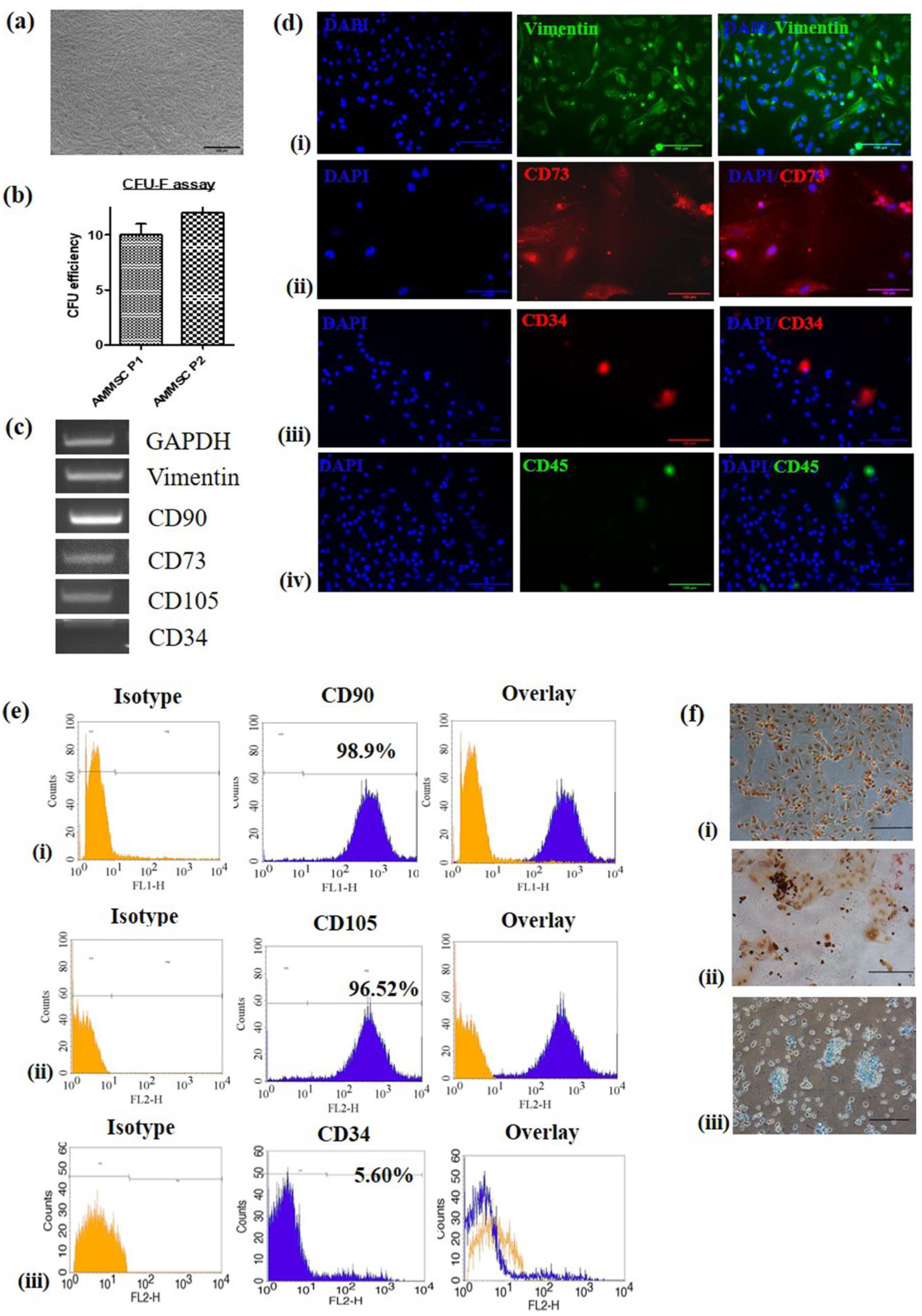
2.1. Isolation, Culture and Characterization of AM-MSC
2.2. Trilineage Differentiation
2.3. Differentiation of AM-MSCs into Neuronal Cells
2.4. AD-like Pathology on Treatment with Aβ1–42
2.5. Wnt Regulators in AD
2.6. Telomerase Activity
2.7. Effect of AD Drugs on the In Vitro AD Model
2.8. Oxidative Stress and Impaired Metabolism
2.9. Aβ1–42 and Wnt Antagonist Induced Neurotoxicity in Mice
2.10. Downregulation of AD201 Significantly Restored the Expression of Neuronal Markers and Wnt Canonical Genes
3. Discussion
4. Methods
4.1. Isolation of MSC from Amniotic Membrane
4.2. Induction of Neurogenesis in AM-MSC
4.3. Cell Viability and Mitochondrial Staining
4.4. Molecular Analysis of Differentiation
4.5. Immunofluorescence and Flow Cytometry
4.6. Induction of Neurodegeneration
4.7. TRAP Assay
4.8. Drug Studies
4.9. Animal Study
4.10. AD201 Silencing Study
4.11. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
List of Abbreviations
References
- DeTure, M.A.; Dickson, D.W. The neuropathological diagnosis of Alzheimer’s disease. Mol. Neurodegener. 2019, 14, 32. [Google Scholar] [CrossRef]
- Vaillant-Beuchot, L.; Mary, A.; Pardossi-Piquard, R.; Bourgeois, A.; Lauritzen, I.; Eysert, F.; Kinoshita, P.F.; Cazareth, J.; Badot, C.; Fragaki, K.; et al. Accumulation of amyloid precursor protein C-terminal fragments triggers mitochondrial structure, function, and mitophagy defects in Alzheimer’s disease models and human brains. Acta Neuropathol. 2021, 141, 39–65. [Google Scholar] [CrossRef]
- Kato, D.; Takahashi, Y.; Iwata, H.; Hatakawa, Y.; Lee, S.H.; Oe, T. Comparative studies for amyloid beta degradation: “Neprilysin vs insulysin”, “monomeric vs aggregate”, and “whole Abeta40 vs. its peptide fragments”. Biochem. Biophys. Rep. 2022, 30, 101268. [Google Scholar] [CrossRef]
- Parr, C.; Mirzaei, N.; Christian, M.; Sastre, M. Activation of the Wnt/beta-catenin pathway represses the transcription of the beta-amyloid precursor protein cleaving enzyme (BACE1) via binding of T-cell factor-4 to BACE1 promoter. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2015, 29, 623–635. [Google Scholar] [CrossRef]
- Purro, S.A.; Dickins, E.M.; Salinas, P.C. The secreted Wnt antagonist Dickkopf-1 is required for amyloid beta-mediated synaptic loss. J. Neurosci. 2012, 32, 3492–3498. [Google Scholar] [CrossRef]
- Killick, R.; Ribe, E.M.; Al-Shawi, R.; Malik, B.; Hooper, C.; Fernandes, C.; Dobson, R.; Nolan, P.M.; Lourdusamy, A.; Furney, S.; et al. Clusterin regulates beta-amyloid toxicity via Dickkopf-1-driven induction of the wnt-PCP-JNK pathway. Mol. Psychiatry 2014, 19, 88–98. [Google Scholar] [CrossRef]
- Slanzi, A.; Iannoto, G.; Rossi, B.; Zenaro, E.; Constantin, G. In vitro Models of Neurodegenerative Diseases. Front. Cell Dev. Biol. 2020, 8, 328. [Google Scholar] [CrossRef]
- Schlachetzki, J.C.; Saliba, S.W.; Oliveira, A.C. Studying neurodegenerative diseases in culture models. Rev. Bras. Psiquiatr. 2013, 35 (Suppl. S2), S92–S100. [Google Scholar] [CrossRef]
- Loo, D.T.; Copani, A.; Pike, C.J.; Whittemore, E.R.; Walencewicz, A.J.; Cotman, C.W. Apoptosis is induced by beta-amyloid in cultured central nervous system neurons. Proc. Natl. Acad. Sci. USA 1993, 90, 7951–7955. [Google Scholar] [CrossRef]
- Mhatre, S.D.; Paddock, B.E.; Saunders, A.J.; Marenda, D.R. Invertebrate models of Alzheimer’s disease. J. Alzheimers Dis. 2013, 33, 3–16. [Google Scholar] [CrossRef]
- Honda, M.; Minami, I.; Tooi, N.; Morone, N.; Nishioka, H.; Uemura, K.; Kinoshita, A.; Heuser, J.E.; Nakatsuji, N.; Aiba, K. The modeling of Alzheimer’s disease by the overexpression of mutant Presenilin 1 in human embryonic stem cells. Biochem. Biophys. Res. Commun. 2016, 469, 587–592. [Google Scholar] [CrossRef]
- Warrier, S.; Haridas, N.; Bhonde, R. Inherent propensity of amnion-derived mesenchymal stem cells towards endothelial lineage: Vascularization from an avascular tissue. Placenta 2012, 33, 850–858. [Google Scholar] [CrossRef]
- Facchinetti, R.; Bronzuoli, M.R.; Scuderi, C. An Animal Model of Alzheimer Disease Based on the Intrahippocampal Injection of Amyloid beta-Peptide (1–42). Methods Mol. Biol. 2018, 1727, 343–352. [Google Scholar] [CrossRef]
- Guzmán, B.C.-F.; Chaffey, T.E.; Palpagama, T.H.; Waters, S.; Boix, J.; Tate, W.P.; Peppercorn, K.; Dragunow, M.; Waldvogel, H.J.; Faull, R.L.M. The interplay between beta-amyloid 1–42 (Aβ1–42)-induced hippocampal inflammatory response, p-tau, vascular pathology, and their synergistic contributions to neuronal death and behavioral deficits. Front. Mol. Neurosci. 2020, 13, 522073. [Google Scholar] [CrossRef]
- Yu, X.; Li, Y.; Mu, X. Effect of Quercetin on PC12 Alzheimer’s Disease Cell Model Induced by Aβ25-35 and Its Mechanism Based on Sirtuin1/Nrf2/HO-1 Pathway. Biomed Res. Int. 2020, 2020, 8210578. [Google Scholar] [CrossRef]
- Krishtal, J.; Bragina, O.; Metsla, K.; Palumaa, P.; Tõugu, V. In situ fibrillizing amyloid-beta 1-42 induces neurite degeneration and apoptosis of differentiated SH-SY5Y cells. PLoS ONE 2017, 12, e0186636. [Google Scholar] [CrossRef]
- Park, D.; Yang, Y.-H.; Bae, D.K.; Lee, S.H.; Yang, G.; Kyung, J.; Kim, D.; Choi, E.-K.; Lee, S.W.; Kim, G.H. Improvement of cognitive function and physical activity of aging mice by human neural stem cells over-expressing choline acetyltransferase. Neurobiol. Aging 2013, 34, 2639–2646. [Google Scholar] [CrossRef]
- Divya, M.S.; Roshin, G.E.; Divya, T.S.; Rasheed, V.A.; Santhoshkumar, T.R.; Elizabeth, K.E.; James, J.; Pillai, R.M. Umbilical cord blood-derived mesenchymal stem cells consist of a unique population of progenitors co-expressing mesenchymal stem cell and neuronal markers capable of instantaneous neuronal differentiation. Stem Cell Res. Ther. 2012, 3, 57. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.B.; Lindholm, K.; Yan, R.; Citron, M.; Xia, W.; Yang, X.L.; Beach, T.; Sue, L.; Wong, P.; Price, D.; et al. Elevated beta-secretase expression and enzymatic activity detected in sporadic Alzheimer disease. Nat. Med. 2003, 9, 3–4. [Google Scholar] [CrossRef]
- Hess, D.C.; Borlongan, C. Stem cells and neurological diseases. Cell Prolif. 2008, 41, 94–114. [Google Scholar] [CrossRef] [PubMed]
- Auld, D.S.; Mennicken, F.; Quirion, R. Nerve growth factor rapidly induces prolonged acetylcholine release from cultured basal forebrain neurons: Differentiation between neuromodulatory and neurotrophic influences. J. Neurosci. 2001, 21, 3375–3382. [Google Scholar] [CrossRef] [PubMed]
- Åberg, M.A.; Åberg, N.D.; Hedbäcker, H.; Oscarsson, J.; Eriksson, P.S. Peripheral infusion of IGF-I selectively induces neurogenesis in the adult rat hippocampus. J. Neurosci. 2000, 20, 2896–2903. [Google Scholar] [CrossRef] [PubMed]
- Crigler, L.; Robey, R.C.; Asawachaicharn, A.; Gaupp, D.; Phinney, D.G. Human mesenchymal stem cell subpopulations express a variety of neuro-regulatory molecules and promote neuronal cell survival and neuritogenesis. Exp. Neurol. 2006, 198, 54–64. [Google Scholar] [CrossRef] [PubMed]
- George, S.; Hamblin, M.R.; Abrahamse, H. Differentiation of Mesenchymal Stem Cells to Neuroglia: In the Context of Cell Signalling. Stem Cell Rev. Rep. 2019, 15, 814–826. [Google Scholar] [CrossRef] [PubMed]
- Jang, S.; Cho, H.-H.; Cho, Y.-B.; Park, J.-S.; Jeong, H.-S. Functional neural differentiation of human adipose tissue-derived stem cells using bFGF and forskolin. BMC Cell Biol. 2010, 11, 25. [Google Scholar] [CrossRef]
- Rooney, G.E.; Howard, L.; O’Brien, T.; Windebank, A.J.; Barry, F.P. Elevation of cAMP in mesenchymal stem cells transiently upregulates neural markers rather than inducing neural differentiation. Stem Cells Dev. 2009, 18, 387–398. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.Y.; Lee, D.H.; D’Andrea, M.R.; Peterson, P.A.; Shank, R.P.; Reitz, A.B. beta-Amyloid(1–42) binds to alpha7 nicotinic acetylcholine receptor with high affinity. Implications for Alzheimer’s disease pathology. J. Biol. Chem. 2000, 275, 5626–5632. [Google Scholar] [CrossRef]
- Stancu, I.C.; Vasconcelos, B.; Terwel, D.; Dewachter, I. Models of beta-amyloid induced Tau-pathology: The long and “folded” road to understand the mechanism. Mol. Neurodegener. 2014, 9, 51. [Google Scholar] [CrossRef]
- Kim, Y.S.; Jung, H.M.; Yoon, B.E. Exploring glia to better understand Alzheimer’s disease. Anim. Cells Syst. (Seoul) 2018, 22, 213–218. [Google Scholar] [CrossRef]
- Tyurikova, O.; Zheng, K.; Rings, A.; Drews, A.; Klenerman, D.; Rusakov, D.A. Monitoring Ca2+ elevations in individual astrocytes upon local release of amyloid beta in acute brain slices. Brain Res. Bull. 2018, 136, 85–90. [Google Scholar] [CrossRef] [PubMed]
- Whitehouse, P.J.; Price, D.L.; Struble, R.G.; Clark, A.W.; Coyle, J.T.; Delon, M.R. Alzheimer’s disease and senile dementia: Loss of neurons in the basal forebrain. Science 1982, 215, 1237–1239. [Google Scholar] [CrossRef]
- Bell, K.F.; Ducatenzeiler, A.; Ribeiro-da-Silva, A.; Duff, K.; Bennett, D.A.; Cuello, A.C. The amyloid pathology progresses in a neurotransmitter-specific manner. Neurobiol. Aging 2006, 27, 1644–1657. [Google Scholar] [CrossRef]
- Shen, J.; Wu, J. Nicotinic cholinergic mechanisms in Alzheimer’s disease. Int. Rev. Neurobiol. 2015, 124, 275–292. [Google Scholar]
- Fukumoto, H.; Cheung, B.S.; Hyman, B.T.; Irizarry, M.C. Beta-secretase protein and activity are increased in the neocortex in Alzheimer disease. Arch. Neurol. 2002, 59, 1381–1389. [Google Scholar] [CrossRef]
- Li, R.; Lindholm, K.; Yang, L.B.; Yue, X.; Citron, M.; Yan, R.; Beach, T.; Sue, L.; Sabbagh, M.; Cai, H.; et al. Amyloid beta peptide load is correlated with increased beta-secretase activity in sporadic Alzheimer’s disease patients. Proc. Natl. Acad. Sci. USA 2004, 101, 3632–3637. [Google Scholar] [CrossRef]
- Babusikova, E.; Dobrota, D.; Turner, A.J.; Nalivaeva, N.N. Effect of Global Brain Ischemia on Amyloid Precursor Protein Metabolism and Expression of Amyloid-Degrading Enzymes in Rat Cortex: Role in Pathogenesis of Alzheimer’s Disease. Biochemistry 2021, 86, 680–692. [Google Scholar] [CrossRef]
- Sahoo, B.R.; Panda, P.K.; Liang, W.; Tang, W.J.; Ahuja, R.; Ramamoorthy, A. Degradation of Alzheimer’s Amyloid-beta by a Catalytically Inactive Insulin-Degrading Enzyme. J. Mol. Biol. 2021, 433, 166993. [Google Scholar] [CrossRef]
- Nalivaeva, N.N.; Turner, A.J. Targeting amyloid clearance in Alzheimer’s disease as a therapeutic strategy. Br. J. Pharm. 2019, 176, 3447–3463. [Google Scholar] [CrossRef]
- Mawuenyega, K.G.; Sigurdson, W.; Ovod, V.; Munsell, L.; Kasten, T.; Morris, J.C.; Yarasheski, K.E.; Bateman, R.J. Decreased clearance of CNS beta-amyloid in Alzheimer’s disease. Science 2010, 330, 1774. [Google Scholar] [CrossRef]
- Palop, J.J.; Mucke, L. Amyloid-β–induced neuronal dysfunction in Alzheimer’s disease: From synapses toward neural networks. Nat. Neurosci. 2010, 13, 812. [Google Scholar] [CrossRef]
- Fifre, A.; Sponne, I.; Koziel, V.; Kriem, B.; Potin, F.T.Y.; Bihain, B.E.; Olivier, J.-L.; Oster, T.; Pillot, T. Microtubule-associated protein MAP1A, MAP1B, and MAP2 proteolysis during soluble amyloid β-peptide-induced neuronal apoptosis: Synergistic involvement of calpain and caspase-3. J. Biol. Chem. 2006, 281, 229–240. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Dong, X.P. Dysfunction of microtubule-associated proteins of MAP2/tau family in Prion disease. Prion 2012, 6, 334–338. [Google Scholar] [CrossRef]
- Xie, C.; Miyasaka, T.; Yoshimura, S.; Hatsuta, H.; Yoshina, S.; Kage-Nakadai, E.; Mitani, S.; Murayama, S.; Ihara, Y. The homologous carboxyl-terminal domains of microtubule-associated protein 2 and TAU induce neuronal dysfunction and have differential fates in the evolution of neurofibrillary tangles. PLoS ONE 2014, 9, e89796. [Google Scholar] [CrossRef]
- Kandimalla, R.; Manczak, M.; Yin, X.; Wang, R.; Reddy, P.H. Hippocampal phosphorylated tau induced cognitive decline, dendritic spine loss and mitochondrial abnormalities in a mouse model of Alzheimer’s disease. Hum. Mol. Genet. 2018, 27, 30–40. [Google Scholar] [CrossRef]
- Li, B.; Yamamori, H.; Tatebayashi, Y.; Shafit-Zagardo, B.; Tanimukai, H.; Chen, S.; Iqbal, K.; Grundke-Iqbal, I. Failure of neuronal maturation in Alzheimer disease dentate gyrus. J. Neuropathol. Exp. Neurol. 2008, 67, 78–84. [Google Scholar] [CrossRef]
- Iovino, M.; Pfisterer, U.; Holton, J.L.; Lashley, T.; Swingler, R.J.; Calo, L.; Treacy, R.; Revesz, T.; Parmar, M.; Goedert, M. The novel MAPT mutation K298E: Mechanisms of mutant tau toxicity, brain pathology and tau expression in induced fibroblast-derived neurons. Acta Neuropathol. 2014, 127, 283–295. [Google Scholar] [CrossRef]
- Oliveira, J.; Costa, M.; de Almeida, M.S.C.; da Cruz, E.S.O.A.B.; Henriques, A.G. Protein Phosphorylation is a Key Mechanism in Alzheimer’s Disease. J. Alzheimers Dis. 2017, 58, 953–978. [Google Scholar] [CrossRef] [PubMed]
- Liao, H.; Li, Y.; Brautigan, D.L.; Gundersen, G.G. Protein phosphatase 1 is targeted to microtubules by the microtubule-associated protein Tau. J. Biol. Chem. 1998, 273, 21901–21908. [Google Scholar] [CrossRef]
- Sontag, E.; Nunbhakdi-Craig, V.; Lee, G.; Brandt, R.; Kamibayashi, C.; Kuret, J.; White, C.L., 3rd; Mumby, M.C.; Bloom, G.S. Molecular interactions among protein phosphatase 2A, tau, and microtubules. Implications for the regulation of tau phosphorylation and the development of tauopathies. J. Biol. Chem. 1999, 274, 25490–25498. [Google Scholar] [CrossRef]
- Sun, W.; Qureshi, H.Y.; Cafferty, P.W.; Sobue, K.; Agarwal-Mawal, A.; Neufield, K.D.; Paudel, H.K. Glycogen synthase kinase-3beta is complexed with tau protein in brain microtubules. J. Biol. Chem. 2002, 277, 11933–11940. [Google Scholar] [CrossRef]
- Sinsky, J.; Majerova, P.; Kovac, A.; Kotlyar, M.; Jurisica, I.; Hanes, J. Physiological Tau Interactome in Brain and Its Link to Tauopathies. J. Proteome Res. 2020, 19, 2429–2442. [Google Scholar] [CrossRef] [PubMed]
- Hooper, C.; Killick, R.; Lovestone, S. The GSK3 hypothesis of Alzheimer’s disease. J. Neurochem. 2008, 104, 1433–1439. [Google Scholar] [CrossRef] [PubMed]
- Keeney, J.T.; Swomley, A.M.; Harris, J.L.; Fiorini, A.; Mitov, M.I.; Perluigi, M.; Sultana, R.; Butterfield, D.A. Cell cycle proteins in brain in mild cognitive impairment: Insights into progression to Alzheimer disease. Neurotox Res. 2012, 22, 220–230. [Google Scholar] [CrossRef] [PubMed]
- Lee, G.; Thangavel, R.; Sharma, V.M.; Litersky, J.M.; Bhaskar, K.; Fang, S.M.; Do, L.H.; Andreadis, A.; Van Hoesen, G.; Ksiezak-Reding, H. Phosphorylation of tau by fyn: Implications for Alzheimer’s disease. J. Neurosci. 2004, 24, 2304–2312. [Google Scholar] [CrossRef] [PubMed]
- Sharma, V.M.; Litersky, J.M.; Bhaskar, K.; Lee, G. Tau impacts on growth-factor-stimulated actin remodeling. J. Cell Sci. 2007, 120, 748–757. [Google Scholar] [CrossRef]
- Ittner, L.M.; Ke, Y.D.; Delerue, F.; Bi, M.; Gladbach, A.; van Eersel, J.; Wolfing, H.; Chieng, B.C.; Christie, M.J.; Napier, I.A.; et al. Dendritic function of tau mediates amyloid-beta toxicity in Alzheimer’s disease mouse models. Cell 2010, 142, 387–397. [Google Scholar] [CrossRef]
- Nakazawa, T.; Komai, S.; Tezuka, T.; Hisatsune, C.; Umemori, H.; Semba, K.; Mishina, M.; Manabe, T.; Yamamoto, T. Characterization of Fyn-mediated tyrosine phosphorylation sites on GluR epsilon 2 (NR2B) subunit of the N-methyl-D-aspartate receptor. J. Biol. Chem. 2001, 276, 693–699. [Google Scholar] [CrossRef]
- Roche, K.W.; Standley, S.; McCallum, J.; Dune Ly, C.; Ehlers, M.D.; Wenthold, R.J. Molecular determinants of NMDA receptor internalization. Nat. Neurosci. 2001, 4, 794–802. [Google Scholar] [CrossRef]
- Wang, P.; Guan, P.P.; Guo, J.W.; Cao, L.L.; Xu, G.B.; Yu, X.; Wang, Y.; Wang, Z.Y. Prostaglandin I2 upregulates the expression of anterior pharynx-defective-1alpha and anterior pharynx-defective-1beta in amyloid precursor protein/presenilin 1 transgenic mice. Aging Cell 2016, 15, 861–871. [Google Scholar] [CrossRef]
- Qin, J.; Zhang, X.; Wang, Z.; Li, J.; Zhang, Z.; Gao, L.; Ren, H.; Qian, M.; Du, B. Presenilin 2 deficiency facilitates Abeta-induced neuroinflammation and injury by upregulating P2X7 expression. Sci. China Life Sci. 2017, 60, 189–201. [Google Scholar] [CrossRef]
- Delabio, R.; Rasmussen, L.; Mizumoto, I.; Viani, G.A.; Chen, E.; Villares, J.; Costa, I.B.; Turecki, G.; Linde, S.A.; Smith, M.C.; et al. PSEN1 and PSEN2 gene expression in Alzheimer’s disease brain: A new approach. J. Alzheimers Dis. 2014, 42, 757–760. [Google Scholar] [CrossRef]
- Logan, C.Y.; Nusse, R. The Wnt signaling pathway in development and disease. Annu. Rev. Cell Dev. Biol. 2004, 20, 781–810. [Google Scholar] [CrossRef]
- Speese, S.D.; Budnik, V. Wnts: Up-and-coming at the synapse. Trends Neurosci. 2007, 30, 268–275. [Google Scholar] [CrossRef] [PubMed]
- Wan, W.; Xia, S.; Kalionis, B.; Liu, L.; Li, Y. The role of Wnt signaling in the development of alzheimer’s disease: A potential therapeutic target? Biomed Res. Int. 2014, 2014, 301575. [Google Scholar] [CrossRef] [PubMed]
- Tapia-Rojas, C.; Inestrosa, N.C. Loss of canonical Wnt signaling is involved in the pathogenesis of Alzheimer’s disease. Neural. Regen. Res. 2018, 13, 1705. [Google Scholar]
- Jia, L.; Piña-Crespo, J.; Li, Y. Restoring Wnt/β-catenin signaling is a promising therapeutic strategy for Alzheimer’s disease. Mol. Brain 2019, 12, 104. [Google Scholar] [CrossRef] [PubMed]
- Rankin, C.A.; Sun, Q.; Gamblin, T.C. Tau phosphorylation by GSK-3beta promotes tangle-like filament morphology. Mol. Neurodegener. 2007, 2, 12. [Google Scholar] [CrossRef]
- Caricasole, A.; Copani, A.; Caraci, F.; Aronica, E.; Rozemuller, A.J.; Caruso, A.; Storto, M.; Gaviraghi, G.; Terstappen, G.C.; Nicoletti, F. Induction of Dickkopf-1, a negative modulator of the Wnt pathway, is associated with neuronal degeneration in Alzheimer’s brain. J. Neurosci. 2004, 24, 6021–6027. [Google Scholar] [CrossRef]
- Rosi, M.C.; Luccarini, I.; Grossi, C.; Fiorentini, A.; Spillantini, M.G.; Prisco, A.; Scali, C.; Gianfriddo, M.; Caricasole, A.; Terstappen, G.C. Increased Dickkopf-1 expression in transgenic mouse models of neurodegenerative disease. J. Neurochem. 2010, 112, 1539–1551. [Google Scholar] [CrossRef]
- Zhang, Q.G.; Wang, R.; Khan, M.; Mahesh, V.; Brann, D.W. Role of Dickkopf-1, an antagonist of the Wnt/beta-catenin signaling pathway, in estrogen-induced neuroprotection and attenuation of tau phosphorylation. J. Neurosci. 2008, 28, 8430–8441. [Google Scholar] [CrossRef]
- Inestrosa, N.C.; Godoy, J.A.; Vargas, J.Y.; Arrazola, M.S.; Rios, J.A.; Carvajal, F.J.; Serrano, F.G.; Farias, G.G. Nicotine prevents synaptic impairment induced by amyloid-beta oligomers through alpha7-nicotinic acetylcholine receptor activation. Neuromol. Med. 2013, 15, 549–569. [Google Scholar] [CrossRef] [PubMed]
- Shankar, G.M.; Bloodgood, B.L.; Townsend, M.; Walsh, D.M.; Selkoe, D.J.; Sabatini, B.L. Natural oligomers of the Alzheimer amyloid-β protein induce reversible synapse loss by modulating an NMDA-type glutamate receptor-dependent signaling pathway. J. Neurosci. 2007, 27, 2866–2875. [Google Scholar] [CrossRef] [PubMed]
- Kuchibhotla, K.V.; Goldman, S.T.; Lattarulo, C.R.; Wu, H.-Y.; Hyman, B.T.; Bacskai, B.J. Aβ plaques lead to aberrant regulation of calcium homeostasis in vivo resulting in structural and functional disruption of neuronal networks. Neuron 2008, 59, 214–225. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.-Y.; Hudry, E.; Hashimoto, T.; Kuchibhotla, K.; Rozkalne, A.; Fan, Z.; Spires-Jones, T.; Xie, H.; Arbel-Ornath, M.; Grosskreutz, C.L. Amyloid β induces the morphological neurodegenerative triad of spine loss, dendritic simplification, and neuritic dystrophies through calcineurin activation. J. Neurosci. 2010, 30, 2636–2649. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, N.; Schohl, A.; Ruthazer, E.S. Neural activity regulates synaptic properties and dendritic structure in vivo through calcineurin/NFAT signaling. Neuron 2009, 62, 655–669. [Google Scholar] [CrossRef] [PubMed]
- Reese, L.C.; Taglialatela, G. Neuroimmunomodulation by calcineurin in aging and Alzheimer’s disease. Aging Dis. 2010, 1, 245–253. [Google Scholar] [PubMed]
- Hudry, E.; Wu, H.-Y.; Arbel-Ornath, M.; Hashimoto, T.; Matsouaka, R.; Fan, Z.; Spires-Jones, T.L.; Betensky, R.A.; Bacskai, B.J.; Hyman, B.T. Inhibition of the NFAT pathway alleviates amyloid beta neurotoxicity in a mouse model of Alzheimer’s disease. J. Neurosci. 2012, 32, 3176–3192. [Google Scholar] [CrossRef]
- Landeira, B.S.; Santana, T.T.d.S.; Araújo, J.A.d.M.; Tabet, E.I.; Tannous, B.A.; Schroeder, T.; Costa, M.R. Activity-independent effects of CREB on neuronal survival and differentiation during mouse cerebral cortex development. Cereb Cortex 2018, 28, 538–548. [Google Scholar] [CrossRef]
- Ghosh, A.; Giese, K.P. Calcium/calmodulin-dependent kinase II and Alzheimer’s disease. Mol. Brain 2015, 8, 78. [Google Scholar] [CrossRef]
- Ibanez, K.; Boullosa, C.; Tabares-Seisdedos, R.; Baudot, A.; Valencia, A. Molecular evidence for the inverse comorbidity between central nervous system disorders and cancers detected by transcriptomic meta-analyses. PLoS Genet. 2014, 10, e1004173. [Google Scholar] [CrossRef]
- Bhuvanalakshmi, G.; Gamit, N.; Patil, M.; Arfuso, F.; Sethi, G.; Dharmarajan, A.; Kumar, A.P.; Warrier, S. Stemness, Pluripotentiality, and Wnt Antagonism: sFRP4, a Wnt antagonist Mediates Pluripotency and Stemness in Glioblastoma. Cancers 2018, 11, 25. [Google Scholar] [CrossRef]
- McCubrey, J.A.; Steelman, L.S.; Bertrand, F.E.; Davis, N.M.; Sokolosky, M.; Abrams, S.L.; Montalto, G.; D’Assoro, A.B.; Libra, M.; Nicoletti, F.; et al. GSK-3 as potential target for therapeutic intervention in cancer. Oncotarget 2014, 5, 2881–2911. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sayas, C.L.; Avila, J. GSK-3 and Tau: A Key Duet in Alzheimer’s Disease. Cells 2021, 10, 721. [Google Scholar] [CrossRef]
- Okino, K.; Nagai, H.; Hatta, M.; Nagahata, T.; Yoneyama, K.; Ohta, Y.; Jin, E.; Kawanami, O.; Araki, T.; Emi, M. Up-regulation and overproduction of DVL-1, the human counterpart of the Drosophila dishevelled gene, in cervical squamous cell carcinoma. Oncol. Rep. 2003, 10, 1219–1223. [Google Scholar] [CrossRef]
- Uematsu, K.; He, B.; You, L.; Xu, Z.; McCormick, F.; Jablons, D.M. Activation of the Wnt pathway in non small cell lung cancer: Evidence of dishevelled overexpression. Oncogene 2003, 22, 7218–7221. [Google Scholar] [CrossRef]
- Uematsu, K.; Kanazawa, S.; You, L.; He, B.; Xu, Z.; Li, K.; Peterlin, B.M.; McCormick, F.; Jablons, D.M. Wnt pathway activation in mesothelioma: Evidence of Dishevelled overexpression and transcriptional activity of beta-catenin. Cancer Res. 2003, 63, 4547–4551. [Google Scholar]
- Dou, Y.; Kawaler, E.A.; Cui Zhou, D.; Gritsenko, M.A.; Huang, C.; Blumenberg, L.; Karpova, A.; Petyuk, V.A.; Savage, S.R.; Satpathy, S.; et al. Proteogenomic Characterization of Endometrial Carcinoma. Cell 2020, 180, 729–748.e26. [Google Scholar] [CrossRef]
- Vyas, C.M.; Ogata, S.; Reynolds, C.F.; Mischoulon, D.; Chang, G.; Cook, N.R.; Manson, J.E.; Crous-Bou, M.; De Vivo, I.; Okereke, O.I. Telomere length and its relationships with lifestyle and behavioural factors: Variations by sex and race/ethnicity. Age Ageing 2021, 50, 838–846. [Google Scholar] [CrossRef]
- Fernandes, S.G.; Dsouza, R.; Khattar, E. External environmental agents influence telomere length and telomerase activity by modulating internal cellular processes: Implications in human aging. Environ. Toxicol. Pharm. 2021, 85, 103633. [Google Scholar] [CrossRef]
- Boccardi, V.; Paolisso, G.; Mecocci, P. Nutrition and lifestyle in healthy aging: The telomerase challenge. Aging (Albany NY) 2016, 8, 12–15. [Google Scholar] [CrossRef]
- Gavia-Garcia, G.; Rosado-Perez, J.; Arista-Ugalde, T.L.; Aguiniga-Sanchez, I.; Santiago-Osorio, E.; Mendoza-Nunez, V.M. Telomere Length and Oxidative Stress and Its Relation with Metabolic Syndrome Components in the Aging. Biology 2021, 10, 253. [Google Scholar] [CrossRef]
- Yadav, S.; Maurya, P.K. Correlation Between Telomere Length and Biomarkers of Oxidative Stress in Human Aging. Rejuvenation Res. 2022, 25, 25–29. [Google Scholar] [CrossRef] [PubMed]
- Rossiello, F.; Jurk, D.; Passos, J.F.; d’Adda di Fagagna, F. Telomere dysfunction in ageing and age-related diseases. Nat. Cell Biol. 2022, 24, 135–147. [Google Scholar] [CrossRef] [PubMed]
- Spilsbury, A.; Miwa, S.; Attems, J.; Saretzki, G. The role of telomerase protein TERT in Alzheimer’s disease and in tau-related pathology in vitro. J. Neurosci. 2015, 35, 1659–1674. [Google Scholar] [CrossRef]
- Haendeler, J.; Dröse, S.; Büchner, N.; Jakob, S.; Altschmied, J.; Goy, C.; Spyridopoulos, I.; Zeiher, A.M.; Brandt, U.; Dimmeler, S. Mitochondrial telomerase reverse transcriptase binds to and protects mitochondrial DNA and function from damage. Arter. Thromb. Vasc. Biol. 2009, 29, 929–935. [Google Scholar] [CrossRef]
- Campanari, M.-L.; García-Ayllón, M.-S.; Blazquez-Llorca, L.; Luk, W.K.; Tsim, K.; Sáez-Valero, J. Acetylcholinesterase protein level is preserved in the Alzheimer’s brain. J. Mol. Neurosci. 2014, 53, 446–453. [Google Scholar] [CrossRef] [PubMed]
- Lane, R.M.; Potkin, S.G.; Enz, A. Targeting acetylcholinesterase and butyrylcholinesterase in dementia. Int. J. Neuropsychopharmacol. 2006, 9, 101–124. [Google Scholar] [CrossRef] [PubMed]
- Paul, R.; Borah, A. Global loss of acetylcholinesterase activity with mitochondrial complexes inhibition and inflammation in brain of hypercholesterolemic mice. Sci. Rep. 2017, 7, 17922. [Google Scholar] [CrossRef]
- Sayre, L.M.; Zelasko, D.A.; Harris, P.L.; Perry, G.; Salomon, R.G.; Smith, M.A. 4-Hydroxynonenal-derived advanced lipid peroxidation end products are increased in Alzheimer’s disease. J. Neurochem. 1997, 68, 2092–2097. [Google Scholar] [CrossRef]
- Montine, K.S.; Reich, E.; Neely, M.D.; Sidell, K.R.; Olson, S.J.; Markesbery, W.R.; Montine, T.J. Distribution of reducible 4-hydroxynonenal adduct immunoreactivity in Alzheimer disease is associated with APOE genotype. J. Neuropathol. Exp. Neurol. 1998, 57, 415–425. [Google Scholar] [CrossRef]
- Smith, C.; Carney, J.M.; Starke-Reed, P.; Oliver, C.; Stadtman, E.; Floyd, R.; Markesbery, W. Excess brain protein oxidation and enzyme dysfunction in normal aging and in Alzheimer disease. Proc. Natl. Acad. Sci. USA 1991, 88, 10540–10543. [Google Scholar] [CrossRef] [PubMed]
- Mecocci, P.; MacGarvey, U.; Beal, M.F. Oxidative damage to mitochondrial DNA is increased in Alzheimer’s disease. Ann. Neurol. 1994, 36, 747–751. [Google Scholar] [CrossRef] [PubMed]
- Gabbita, S.P.; Lovell, M.A.; Markesbery, W.R. Increased nuclear DNA oxidation in the brain in Alzheimer’s disease. J. Neurochem. 1998, 71, 2034–2040. [Google Scholar] [CrossRef] [Green Version]
- Goschorska, M.; Gutowska, I.; Baranowska-Bosiacka, I.; Piotrowska, K.; Metryka, E.; Safranow, K.; Chlubek, D. Influence of acetylcholinesterase inhibitors used in Alzheimer’s Disease treatment on the activity of antioxidant enzymes and the concentration of glutathione in THP-1 macrophages under fluoride-induced oxidative stress. Int. J. Environ. Res. Public Health 2019, 16, 10. [Google Scholar] [CrossRef]
- Ross, J.M.; Öberg, J.; Brené, S.; Coppotelli, G.; Terzioglu, M.; Pernold, K.; Goiny, M.; Sitnikov, R.; Kehr, J.; Trifunovic, A. High brain lactate is a hallmark of aging and caused by a shift in the lactate dehydrogenase A/B ratio. Proc. Natl. Acad. Sci. USA 2010, 107, 20087–20092. [Google Scholar] [CrossRef]
- Tapia-Rojas, C.; Inestrosa, N.C. Wnt signaling loss accelerates the appearance of neuropathological hallmarks of Alzheimer’s disease in J20-APP transgenic and wild-type mice. J. Neurochem. 2018, 144, 443–465. [Google Scholar] [CrossRef]
- Dominici, M.; Le Blanc, K.; Mueller, I.; Slaper-Cortenbach, I.; Marini, F.; Krause, D.; Deans, R.; Keating, A.; Prockop, D.; Horwitz, E. Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement. Cytotherapy 2006, 8, 315–317. [Google Scholar] [CrossRef]
- Mandal, S.; Gamit, N.; Varier, L.; Dharmarajan, A.; Warrier, S. Inhibition of breast cancer stem-like cells by a triterpenoid, ursolic acid, via activation of Wnt antagonist, sFRP4 and suppression of miRNA-499a-5p. Life Sci. 2021, 265, 118854. [Google Scholar] [CrossRef]
- Arrazola, M.S.; Ramos-Fernandez, E.; Cisternas, P.; Ordenes, D.; Inestrosa, N.C. Wnt Signaling Prevents the Abeta Oligomer-Induced Mitochondrial Permeability Transition Pore Opening Preserving Mitochondrial Structure in Hippocampal Neurons. PLoS ONE 2017, 12, e0168840. [Google Scholar] [CrossRef]
- Goldblum, D.; Gygax, M.; Bohnke, M.; Garweg, J.G. In vitro toxicity of rivastigmine and donepezil in cells of epithelial origin. Ophthalmic Res. 2002, 34, 97–103. [Google Scholar] [CrossRef]
- Volbracht, C.; van Beek, J.; Zhu, C.; Blomgren, K.; Leist, M. Neuroprotective properties of memantine in different in vitro and in vivo models of excitotoxicity. Eur. J. Neurosci. 2006, 23, 2611–2622. [Google Scholar] [CrossRef] [PubMed]
- Costanzo, P.; Cariati, L.; Desiderio, D.; Sgammato, R.; Lamberti, A.; Arcone, R.; Salerno, R.; Nardi, M.; Masullo, M.; Oliverio, M. Design, Synthesis, and Evaluation of Donepezil-Like Compounds as AChE and BACE-1 Inhibitors. ACS Med. Chem. Lett. 2016, 7, 470–475. [Google Scholar] [CrossRef] [PubMed]
- Akhtar, M.W.; Sanz-Blasco, S.; Dolatabadi, N.; Parker, J.; Chon, K.; Lee, M.S.; Soussou, W.; McKercher, S.R.; Ambasudhan, R.; Nakamura, T.; et al. Elevated glucose and oligomeric beta-amyloid disrupt synapses via a common pathway of aberrant protein S-nitrosylation. Nat. Commun. 2016, 7, 10242. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.; Lu, Y.; Kong, H.; Li, L.; Marshall, C.; Xiao, M.; Ding, J.; Gao, J.; Hu, G. Aquaporin-4 deficiency exacerbates brain oxidative damage and memory deficits induced by long-term ovarian hormone deprivation and D-galactose injection. Int. J. Neuropsychopharmacol. 2012, 15, 55–68. [Google Scholar] [CrossRef]
- Rajamohamedsait, H.B.; Sigurdsson, E.M. Histological staining of amyloid and pre-amyloid peptides and proteins in mouse tissue. Methods Mol. Biol. 2012, 849, 411–424. [Google Scholar] [CrossRef] [PubMed]
- Wilcock, D.M.; Gordon, M.N.; Morgan, D. Quantification of cerebral amyloid angiopathy and parenchymal amyloid plaques with Congo red histochemical stain. Nat. Protoc. 2006, 1, 1591–1595. [Google Scholar] [CrossRef]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Patil, M.; Gamit, N.; Dharmarajan, A.; Sethi, G.; Warrier, S. Identification of a Novel Wnt Antagonist Based Therapeutic and Diagnostic Target for Alzheimer’s Disease Using a Stem Cell-Derived Model. Bioengineering 2023, 10, 192. https://doi.org/10.3390/bioengineering10020192
Patil M, Gamit N, Dharmarajan A, Sethi G, Warrier S. Identification of a Novel Wnt Antagonist Based Therapeutic and Diagnostic Target for Alzheimer’s Disease Using a Stem Cell-Derived Model. Bioengineering. 2023; 10(2):192. https://doi.org/10.3390/bioengineering10020192
Chicago/Turabian StylePatil, Manasi, Naisarg Gamit, Arun Dharmarajan, Gautam Sethi, and Sudha Warrier. 2023. "Identification of a Novel Wnt Antagonist Based Therapeutic and Diagnostic Target for Alzheimer’s Disease Using a Stem Cell-Derived Model" Bioengineering 10, no. 2: 192. https://doi.org/10.3390/bioengineering10020192