
Ageing and Inflammation: What Happens in Periodontium?


Abstract
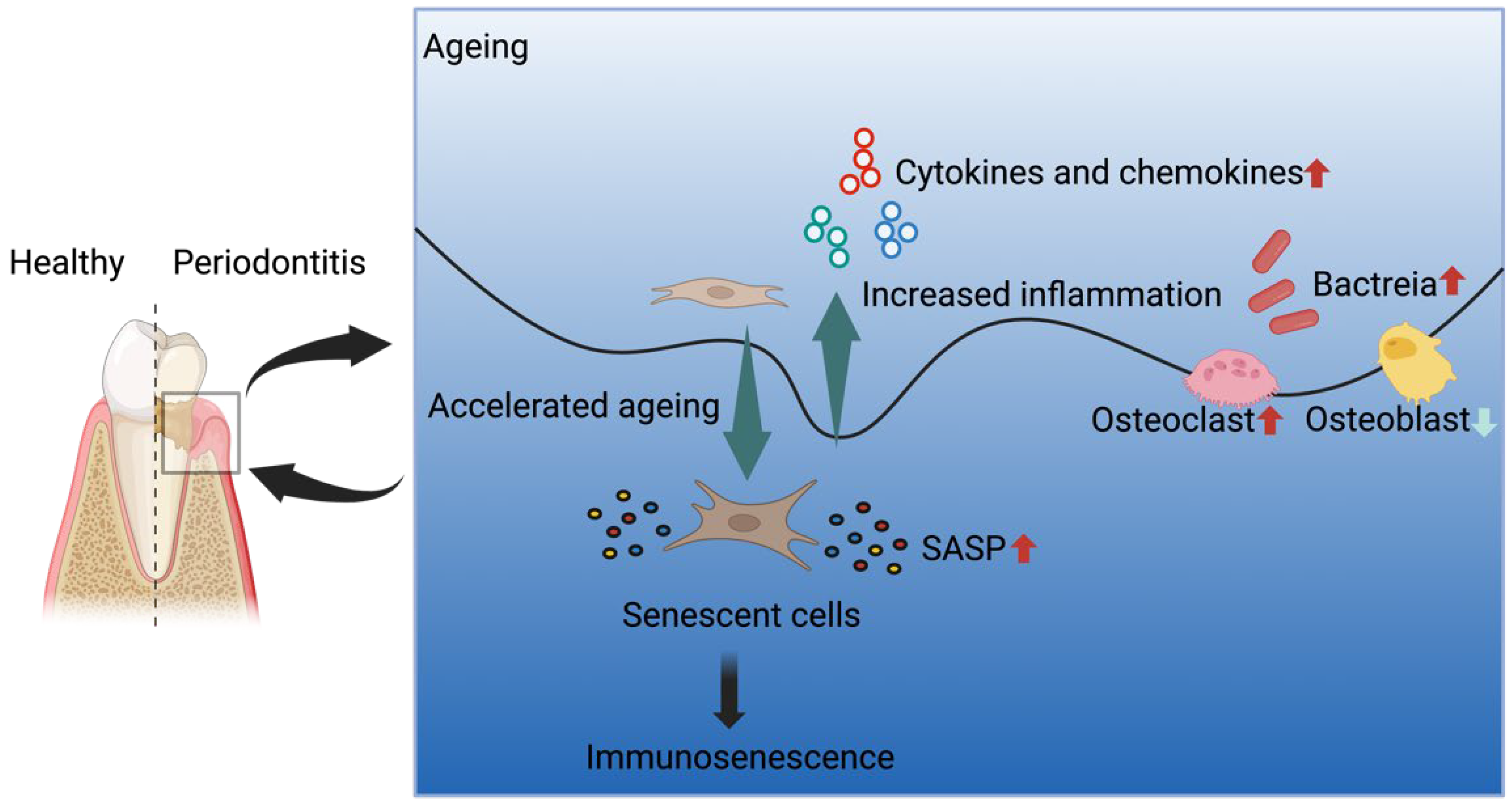
:1. Introduction
2. Ageing and Inflammation
2.1. Inflammaging and Immunosenescence
2.2. Cytokines, Chemokines and Pathways in Ageing and Inflammation
2.2.1. Interleukin-1 Family
2.2.2. Interleukin-6
2.2.3. Tumor Necrosis Factor Alpha
2.2.4. Nuclear Factor-κB (NF-κB) System
3. Periodontitis and Inflammation
3.1. Bacteria in Periodontitis
3.1.1. Lipopolysaccharide (LPS)
3.1.2. Peptidoglycan (PGN)
3.1.3. Gingipains
4. Periodontitis and Ageing
4.1. Cellular Senescence and PDLCs
4.2. Senescence-Associated Secretory Phenotype (SASP) in Periodontitis
4.3. Alveolar Bone Loss in Periodontitis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| CXCL10 | CXC chemokine ligand 10 |
| iNOS | inducible nitric oxide synthase |
| GSK3-β | glycogen synthase kinase 3-β |
| MAPK | mitogen-activated protein kinase |
| PKB | protein kinase B |
References
- Pihlstrom, B.L.; Michalowicz, B.S.; Johnson, N.W. Periodontal diseases. Lancet 2005, 366, 1809–1820. [Google Scholar] [CrossRef] [PubMed]
- Mahendra, J.; Mahendra, L.; Mugri, M.H.; Sayed, M.E.; Bhandi, S.; Alshahrani, R.T.; Balaji, T.M.; Varadarajan, S.; Tanneeru, S.; Srinivasan, S.; et al. Role of periodontal bacteria, viruses, and placental mir155 in chronic periodontitis and Preeclampsia—A genetic microbiological study. Curr. Issues Mol. Biol. 2021, 43, 831–844. [Google Scholar] [CrossRef] [PubMed]
- Wu, R.-X.; Bi, C.-S.; Yu, Y.; Zhang, L.-L.; Chen, F.-M. Age-related decline in the matrix contents and functional properties of human periodontal ligament stem cell sheets. Acta Biomater. 2015, 22, 70–82. [Google Scholar] [CrossRef]
- Chen, F.-M.; Jin, Y. Periodontal tissue engineering and regeneration: Current approaches and expanding opportunities. Tissue Eng. Part B Rev. 2010, 16, 219–255. [Google Scholar] [CrossRef]
- Ebersole, J.L.; Graves, C.L.; Gonzalez, O.A.; Dawson, D., III; Morford, L.A.; Huja, P.E.; Hartsfield, J.K., Jr.; Huja, S.S.; Pandruvada, S.; Wallet, S.M. Aging, inflammation, immunity and periodontal disease. Periodontology 2000 2016, 72, 54–75. [Google Scholar] [CrossRef] [PubMed]
- Franceschi, C.; Campisi, J. Chronic inflammation (inflammaging) and its potential contribution to age-associated diseases. J. Gerontol. Ser. A Biomed. Sci. Med. Sci. 2014, 69, S4–S9. [Google Scholar] [CrossRef]
- Bertl, K.; Tangl, S.; Rybaczek, T.; Berger, B.; Traindl-Prohazka, M.; Schuller-Götzburg, P.; Grossschmidt, K. Prevalence and severity of periodontal disease in a historical Austrian population. J. Periodontal Res. 2020, 55, 931–945. [Google Scholar] [CrossRef]
- Eke, P.I.; Dye, B.; Wei, L.; Thornton-Evans, G.; Genco, R. Prevalence of periodontitis in adults in the United States: 2009 and 2010. J. Dent. Res. 2012, 91, 914–920. [Google Scholar] [CrossRef]
- Tonetti, M.S.; Jepsen, S.; Jin, L.; Otomo-Corgel, J. Impact of the global burden of periodontal diseases on health, nutrition and wellbeing of mankind: A call for global action. J. Clin. Periodontol. 2017, 44, 456–462. [Google Scholar] [CrossRef]
- Huttner, E.A.; Machado, D.C.; De Oliveira, R.B.; Antunes, A.G.F.; Hebling, E. Effects of human aging on periodontal tissues. Spec. Care Dent. 2009, 29, 149–155. [Google Scholar] [CrossRef]
- Guarnieri, R.; Reda, R.; Zanza, A.; Miccoli, G.; Nardo, D.D.; Testarelli, L. Can peri-implant marginal bone loss progression and a-MMP-8 be considered indicators of the subsequent onset of peri-implantitis? A 5-year study. Diagnostics 2022, 12, 2599. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, A.; Banerjee, S.; Wang, Z.; Kong, D.; Majumdar, A.P.; Sarkar, F.H. Aging and inflammation: Etiological culprits of cancer. Curr. Aging Sci. 2009, 2, 174–186. [Google Scholar] [CrossRef] [PubMed]
- Liberale, L.; Montecucco, F.; Schwarz, L.; Lüscher, T.F.; Camici, G.G. Inflammation and cardiovascular diseases: Lessons from seminal clinical trials. Cardiovasc. Res. 2021, 117, 411–422. [Google Scholar] [CrossRef] [PubMed]
- Chung, H.Y.; Kim, D.H.; Lee, E.K.; Chung, K.W.; Chung, S.; Lee, B.; Seo, A.Y.; Chung, J.H.; Jung, Y.S.; Im, E. Redefining chronic inflammation in aging and age-related diseases: Proposal of the senoinflammation concept. Aging Dis. 2019, 10, 367. [Google Scholar] [CrossRef]
- Franceschi, C.; Bonafè, M.; Valensin, S.; Olivieri, F.; De Luca, M.; Ottaviani, E.; De Benedictis, G. Inflamm-aging: An evolutionary perspective on immunosenescence. Ann. N. Y. Acad. Sci. 2000, 908, 244–254. [Google Scholar] [CrossRef]
- Goronzy, J.J.; Weyand, C.M. Successful and maladaptive T cell aging. Immunity 2017, 46, 364–378. [Google Scholar] [CrossRef]
- Pawelec, G. Age and immunity: What is “immunosenescence”? Exp. Gerontol. 2018, 105, 4–9. [Google Scholar] [CrossRef]
- Bruunsgaard, H.; Ladelund, S.; Pedersen, A.N.; Schroll, M.; Jørgensen, T.; Pedersen, B. Predicting death from tumour necrosis factor-alpha and interleukin-6 in 80-year-old people. Clin. Exp. Immunol. 2003, 132, 24–31. [Google Scholar] [CrossRef]
- Kim, H.-J.; Kim, K.-W.; Yu, B.-P.; Chung, H.-Y. The effect of age on cyclooxygenase-2 gene expression: NF-κB activation and IκBα degradation. Free Radic. Biol. Med. 2000, 28, 683–692. [Google Scholar] [CrossRef]
- Kwon, H.J.; Sung, B.K.; Kim, J.W.; Lee, J.H.; Kim, N.D.; Yoo, M.A.; Kang, H.S.; Baek, H.S.; Bae, S.J.; Choi, J.S. The effect of lipopolysaccharide on enhanced inflammatory process with age: Modulation of NF-κB. J. Am. Aging Assoc. 2001, 24, 163–171. [Google Scholar] [CrossRef]
- Ershler, W.B. Interleukin-6: A cytokine for gerontolgists. J. Am. Geriatr. Soc. 1993, 41, 176–181. [Google Scholar] [CrossRef] [PubMed]
- Zanni, F.; Vescovini, R.; Biasini, C.; Fagnoni, F.; Zanlari, L.; Telera, A.; Di Pede, P.; Passeri, G.; Pedrazzoni, M.; Passeri, M. Marked increase with age of type 1 cytokines within memory and effector/cytotoxic CD8+ T cells in humans: A contribution to understand the relationship between inflammation and immunosenescence. Exp. Gerontol. 2003, 38, 981–987. [Google Scholar] [CrossRef] [PubMed]
- Doyle, S.L.; O’Neill, L.A. Toll-like receptors: From the discovery of NFκB to new insights into transcriptional regulations in innate immunity. Biochem. Pharmacol. 2006, 72, 1102–1113. [Google Scholar] [CrossRef] [PubMed]
- Sims, J.E.; Smith, D.E. The IL-1 family: Regulators of immunity. Nat. Rev. Immunol. 2010, 10, 89–102. [Google Scholar] [CrossRef]
- Ferrucci, L.; Harris, T.B.; Guralnik, J.M.; Tracy, R.P.; Corti, M.C.; Cohen, H.J.; Penninx, B.; Pahor, M.; Wallace, R.; Havlik, R.J. Serum IL-6 level and the development of disability in older persons. J. Am. Geriatr. Soc. 1999, 47, 639–646. [Google Scholar] [CrossRef]
- Bruunsgaard, H.; Skinhøj, P.; Pedersen, A.N.; Schroll, M.; Pedersen, B. Ageing, tumour necrosis factor-alpha (TNF-α) and atherosclerosis. Clin. Exp. Immunol. 2000, 121, 255–260. [Google Scholar] [CrossRef]
- Helenius, M.; Kyrylenko, S.; Vehviläinen, P.; Salminen, A. Characterization of aging-associated up-regulation of constitutive nuclear factor-κB binding activity. Antioxid. Redox Signal. 2001, 3, 147–156. [Google Scholar] [CrossRef]
- Akdis, M.; Burgler, S.; Crameri, R.; Eiwegger, T.; Fujita, H.; Gomez, E.; Klunker, S.; Meyer, N.; O’Mahony, L.; Palomares, O. Interleukins, from 1 to 37, and interferon-γ: Receptors, functions, and roles in diseases. J. Allergy Clin. Immunol. 2011, 127, 701–721.e770. [Google Scholar] [CrossRef]
- Forsey, R.; Thompson, J.; Ernerudh, J.; Hurst, T.; Strindhall, J.; Johansson, B.; Nilsson, B.-O.; Wikby, A. Plasma cytokine profiles in elderly humans. Mech. Ageing Dev. 2003, 124, 487–493. [Google Scholar] [CrossRef]
- Wolf, J.; Rose-John, S.; Garbers, C. Interleukin-6 and its receptors: A highly regulated and dynamic system. Cytokine 2014, 70, 11–20. [Google Scholar] [CrossRef]
- Puzianowska-Kuźnicka, M.; Owczarz, M.; Wieczorowska-Tobis, K.; Nadrowski, P.; Chudek, J.; Slusarczyk, P.; Skalska, A.; Jonas, M.; Franek, E.; Mossakowska, M. Interleukin-6 and C-reactive protein, successful aging, and mortality: The PolSenior study. Immun. Ageing 2016, 13, 21. [Google Scholar] [CrossRef] [PubMed]
- Van Epps, P.; Oswald, D.; Higgins, P.; Hornick, T.; Aung, H.; Banks, R.; Wilson, B.; Burant, C.; Gravenstein, S.; Canaday, D. Frailty has a stronger association with inflammation than age in older veterans. Immun. Ageing 2016, 13, 27. [Google Scholar] [CrossRef] [PubMed]
- Palmeri, M.; Misiano, G.; Malaguarnera, M.; Forte, G.I.; Vaccarino, L.; Milano, S.; Scola, L.; Caruso, C.; Motta, M.; Maugeri, D. Cytokine serum profile in a group of Sicilian nonagenarians. J. Immunoass. Immunochem. 2012, 33, 82–90. [Google Scholar] [CrossRef] [PubMed]
- Sansoni, P.; Vescovini, R.; Fagnoni, F.; Biasini, C.; Zanni, F.; Zanlari, L.; Telera, A.; Lucchini, G.; Passeri, G.; Monti, D. The immune system in extreme longevity. Exp. Gerontol. 2008, 43, 61–65. [Google Scholar] [CrossRef]
- Bradley, J. TNF-mediated inflammatory disease. J. Pathol. A J. Pathol. Soc. Great Br. Irel. 2008, 214, 149–160. [Google Scholar] [CrossRef]
- McNerlan, S.; Rea, I.; Alexander, H. A whole blood method for measurement of intracellular TNF-α, IFN-γ and IL-2 expression in stimulated CD3+ lymphocytes: Differences between young and elderly subjects. Exp. Gerontol. 2002, 37, 227–234. [Google Scholar] [CrossRef]
- Armstrong, M.; Alexander, H.; Ritchie, J.; McMillan, S.; Rea, I. Age-related alterations in basal expression and in vitro, tumour necrosis factor alpha mediated, upregulation of CD11b. Gerontology 2001, 47, 180–185. [Google Scholar] [CrossRef]
- Van Den Biggelaar, A.H.; De Craen, A.J.; Gussekloo, J.; Huizinga, T.W.; Heijmans, B.T.; Frölich, M.; Kirkwood, T.B.; Westendorp, R.G. Inflammation underlying cardiovascular mortality is a late consequence of evolutionary programming. FASEB J. 2004, 18, 1022–1024. [Google Scholar] [CrossRef]
- Ross, O.A.; Curran, M.D.; Meenagh, A.; Williams, F.; Barnett, Y.A.; Middleton, D.; Rea, I.M. Study of age-association with cytokine gene polymorphisms in an aged Irish population. Mech. Ageing Dev. 2003, 124, 199–206. [Google Scholar] [CrossRef]
- Lio, D.; Scola, L.; Crivello, A.; Colonna-Romano, G.; Candore, G.; Bonafè, M.; Cavallone, L.; Marchegiani, F.; Olivieri, F.; Franceschi, C. Inflammation, genetics, and longevity: Further studies on the protective effects in men of IL-10− 1082 promoter SNP and its interaction with TNF-α− 308 promoter SNP. J. Med. Genet. 2003, 40, 296–299. [Google Scholar] [CrossRef]
- Cederholm, T.; Persson, M.; Andersson, P.; Stenvinkel, P.; Nordfors, L.; Madden, J.; Vedin, I.; Wretlind, B.; Grimble, R.; Palmblad, J. Polymorphisms in cytokine genes influence long-term survival differently in elderly male and female patients. J. Intern. Med. 2007, 262, 215–223. [Google Scholar] [CrossRef] [PubMed]
- Sako, H.; Omori, K.; Nakayama, M.; Mandai, H.; Ideguchi, H.; Yoshimura-Nakagawa, S.; Sakaida, K.; Nagata-Kamei, C.; Kobayashi, H.; Ishii, S. The Fungal Metabolite (+)-Terrein Abrogates Inflammatory Bone Resorption via the Suppression of TNF-α Production in a Ligature-Induced Periodontitis Mouse Model. J. Fungi 2023, 9, 314. [Google Scholar] [CrossRef] [PubMed]
- Perkins, N.D. Integrating cell-signalling pathways with NF-κB and IKK function. Nat. Rev. Mol. Cell Biol. 2007, 8, 49–62. [Google Scholar] [CrossRef]
- Salminen, A.; Huuskonen, J.; Ojala, J.; Kauppinen, A.; Kaarniranta, K.; Suuronen, T. Activation of innate immunity system during aging: NF-kB signaling is the molecular culprit of inflamm-aging. Ageing Res. Rev. 2008, 7, 83–105. [Google Scholar] [CrossRef] [PubMed]
- Chung, H.; Lee, E.; Choi, Y.; Kim, J.; Kim, D.; Zou, Y.; Kim, C.; Lee, J.; Kim, H.; Kim, N. Molecular inflammation as an underlying mechanism of the aging process and age-related diseases. J. Dent. Res. 2011, 90, 830–840. [Google Scholar] [CrossRef] [PubMed]
- Adler, A.S.; Sinha, S.; Kawahara, T.L.; Zhang, J.Y.; Segal, E.; Chang, H.Y. Motif module map reveals enforcement of aging by continual NF-κB activity. Genes Dev. 2007, 21, 3244–3257. [Google Scholar] [CrossRef]
- Kim, H.-J.; Yu, B.-P.; Chung, H.-Y. Molecular exploration of age-related NF-κB/IKK downregulation by calorie restriction in rat kidney. Free Radic. Biol. Med. 2002, 32, 991–1005. [Google Scholar] [CrossRef]
- Yamashita, T.; Yao, Z.; Li, F.; Zhang, Q.; Badell, I.R.; Schwarz, E.M.; Takeshita, S.; Wagner, E.F.; Noda, M.; Matsuo, K. NF-kappaB p50 and p52 regulate receptor activator of NF-kappaB ligand (RANKL) and tumor necrosis factor-induced osteoclast precursor differentiation by activating c-Fos and NFATc1. J. Biol. Chem. 2007, 282, 18245–18253. [Google Scholar] [CrossRef]
- Huang, X.; Xie, M.; Xie, Y.; Mei, F.; Lu, X.; Li, X.; Chen, L. The roles of osteocytes in alveolar bone destruction in periodontitis. J. Transl. Med. 2020, 18, 479. [Google Scholar] [CrossRef]
- Manolagas, S.C.; Jilka, R.L. Bone marrow, cytokines, and bone remodeling—Emerging insights into the pathophysiology of osteoporosis. N. Engl. J. Med. 1995, 332, 305–311. [Google Scholar] [CrossRef]
- Zaidi, M. Skeletal remodeling in health and disease. Nat. Med. 2007, 13, 791–801. [Google Scholar] [CrossRef]
- Teitelbaum, S.L. Bone resorption by osteoclasts. Science 2000, 289, 1504–1508. [Google Scholar] [CrossRef]
- Teitelbaum, S.L. Osteoclasts: What do they do and how do they do it? Am. J. Pathol. 2007, 170, 427–435. [Google Scholar] [CrossRef]
- Zhou, M.; Graves, D.T. Impact of the host response and osteoblast lineage cells on periodontal disease. Front. Immunol. 2022, 13, 998244. [Google Scholar] [CrossRef] [PubMed]
- Deo, V.; Bhongade, M. Pathogenesis of periodontitis: Role of cytokines in host response. Dent. Today 2010, 29, 60–62, 64. [Google Scholar] [PubMed]
- Nibali, L.; Fedele, S.; D’aiuto, F.; Donos, N. Interleukin-6 in oral diseases: A review. Oral Dis. 2012, 18, 236–243. [Google Scholar] [CrossRef] [PubMed]
- Yucel-Lindberg, T.; Båge, T. Inflammatory mediators in the pathogenesis of periodontitis. Expert Rev. Mol. Med. 2013, 15, e7. [Google Scholar] [CrossRef]
- Bascones, A.; Noronha, S.; Gómez, M.; Mota, P.; Moles, M.; Dorrego, M.V. Tissue destruction in periodontitis: Bacteria or cytokines fault? Quintessence Int. 2005, 36, 299–306. [Google Scholar]
- Huang, G.T.J.; Haake, S.K.; Park, N.H. Gingival epithelial cells increase interleukin-8 secretion in response to Actinobacillus actinomycetemcomitans challenge. J. Periodontol. 1998, 69, 1105–1110. [Google Scholar] [CrossRef]
- Liu, R.K.; Cao, C.F.; Meng, H.X.; Gao, Y. Polymorphonuclear neutrophils and their mediators in gingival tissues from generalized aggressive periodontitis. J. Periodontol. 2001, 72, 1545–1553. [Google Scholar] [CrossRef]
- Gamonal, J.; Acevedo, A.; Bascones, A.; Jorge, O.; Silva, A. Characterization of cellular infiltrate, detection of chemokine receptor CCR5 and interleukin-8 and RANTES chemokines in adult periodontitis. J. Periodontal Res. 2001, 36, 194–203. [Google Scholar] [CrossRef] [PubMed]
- Preshaw, P.M.; Taylor, J.J. How has research into cytokine interactions and their role in driving immune responses impacted our understanding of periodontitis? J. Clin. Periodontol. 2011, 38, 60–84. [Google Scholar] [CrossRef] [PubMed]
- Garlet, G.P.; Martins, W., Jr.; Ferreira, B.R.; Milanezi, C.M.; Silva, J.S. Patterns of chemokines and chemokine receptors expression in different forms of human periodontal disease. J. Periodontal Res. 2003, 38, 210–217. [Google Scholar] [CrossRef]
- Kurtiş, B.; Tüter, G.; Serdar, M.; Akdemir, P.; Uygur, C.; Firatli, E.; Bal, B. Gingival crevicular fluid levels of monocyte chemoattractant protein-1 and tumor necrosis factor-alpha in patients with chronic and aggressive periodontitis. J. Periodontol. 2005, 76, 1849–1855. [Google Scholar] [CrossRef] [PubMed]
- Paster, B.J.; Olsen, I.; Aas, J.A.; Dewhirst, F.E. The breadth of bacterial diversity in the human periodontal pocket and other oral sites. Periodontology 2000 2006, 42, 80–87. [Google Scholar] [CrossRef] [PubMed]
- Rôças, I.N.; Siqueira, J.F., Jr.; Santos, K.R.; Coelho, A.M.; de Janeiro, R. “Red complex”(Bacteroides forsythus, Porphyromonas gingivalis, and Treponema denticola) in endodontic infections: A molecular approach. Oral Surg. Oral Med. Oral Pathol. Oral Radiol. Endodontol. 2001, 91, 468–471. [Google Scholar] [CrossRef]
- Lamont, R.J.; Hajishengallis, G. Polymicrobial synergy and dysbiosis in inflammatory disease. Trends Mol. Med. 2015, 21, 172–183. [Google Scholar] [CrossRef]
- Hajishengallis, G.; Lamont, R.J. Dancing with the stars: How choreographed bacterial interactions dictate nososymbiocity and give rise to keystone pathogens, accessory pathogens, and pathobionts. Trends Microbiol. 2016, 24, 477–489. [Google Scholar] [CrossRef]
- Tomita, S.; Komiya-Ito, A.; Imamura, K.; Kita, D.; Ota, K.; Takayama, S.; Makino-Oi, A.; Kinumatsu, T.; Ota, M.; Saito, A. Prevalence of Aggregatibacter actinomycetemcomitans, Porphyromonas gingivalis and Tannerella forsythia in Japanese patients with generalized chronic and aggressive periodontitis. Microb. Pathog. 2013, 61, 11–15. [Google Scholar] [CrossRef]
- Zhang, W.; Ju, J.; Rigney, T.; Tribble, G. Porphyromonas gingivalis infection increases osteoclastic bone resorption and osteoblastic bone formation in a periodontitis mouse model. BMC Oral Health 2014, 14, 89. [Google Scholar] [CrossRef]
- Wray, D.; Grahame, L. Periodontal bone loss in mice induced by different periodontopathic organisms. Arch. Oral Biol. 1992, 37, 435–438. [Google Scholar] [CrossRef] [PubMed]
- Fine, D.H.; Markowitz, K.; Fairlie, K.; Tischio-Bereski, D.; Ferrendiz, J.; Furgang, D.; Paster, B.J.; Dewhirst, F.E. A consortium of Aggregatibacter actinomycetemcomitans, Streptococcus parasanguinis, and Filifactor alocis is present in sites prior to bone loss in a longitudinal study of localized aggressive periodontitis. J. Clin. Microbiol. 2013, 51, 2850–2861. [Google Scholar] [CrossRef] [PubMed]
- Shaddox, L.; Gonçalves, P.; Vovk, A.; Allin, N.; Huang, H.; Hou, W.; Aukhil, I.; Wallet, S. LPS-induced inflammatory response after therapy of aggressive periodontitis. J. Dent. Res. 2013, 92, 702–708. [Google Scholar] [CrossRef] [PubMed]
- Raetz, C.R.; Whitfield, C. Lipopolysaccharide endotoxins. Annu. Rev. Biochem. 2002, 71, 635–700. [Google Scholar] [CrossRef] [PubMed]
- Jain, S.; Darveau, R.P. Contribution of Porphyromonas gingivalis lipopolysachharide to periodontitis. Periodontology 2000 2010, 54, 53. [Google Scholar] [CrossRef]
- Kukita, A.; Ichigi, Y.; Takigawa, I.; Watanabe, T.; Kukita, T.; Miyamoto, H. Infection of RANKL-primed RAW-D macrophages with Porphyromonas gingivalis promotes osteoclastogenesis in a TNF-α-independent manner. PLoS ONE 2012, 7, e38500. [Google Scholar] [CrossRef]
- Usui, M.; Okamatsu, Y.; Sato, T.; Hanatani, T.; Moritani, Y.; Sano, K.; Yamamoto, M.; Nakashima, K. Thymus-expressed chemokine enhances Porphyromonas gingivalis LPS-induced osteoclast formation via NFATc1 activation. Arch. Oral Biol. 2016, 66, 77–85. [Google Scholar] [CrossRef]
- Jiang, Y.; Mehta, C.K.; Hsu, T.-Y.; Alsulaimani, F.F. Bacteria induce osteoclastogenesis via an osteoblast-independent pathway. Infect. Immun. 2002, 70, 3143–3148. [Google Scholar] [CrossRef]
- Tiranathanagul, S.; Yongchaitrakul, T.; Pattamapun, K.; Pavasant, P. Actinobacillus actinomycetemcomitans lipopolysaccharide activates matrix metalloproteinase-2 and increases receptor activator of nuclear factor-kappaB ligand expression in human periodontal ligament cells. J. Periodontol. 2004, 75, 1647–1654. [Google Scholar] [CrossRef]
- Ali, M.; Kucko, N.; Jansen, J.A.; Yang, F.; Walboomers, X.F. The effect of lipoxin A4 on E. coli LPS-induced osteoclastogenesis. Clin. Oral Investig. 2021, 25, 957–969. [Google Scholar] [CrossRef]
- Rogers, J.E.; Li, F.; Coatney, D.D.; Rossa, C., Jr.; Bronson, P.; Krieder, J.M.; Giannobile, W.V.; Kirkwood, K.L. Actinobacillus actinomycetemcomitans lipopolysaccharide-mediated experimental bone loss model for aggressive periodontitis. J. Periodontol. 2007, 78, 550–558. [Google Scholar] [CrossRef] [PubMed]
- Oka, H.; Miyauchi, M.; Furusho, H.; Nishihara, T.; Takata, T. Oral administration of prostaglandin E2-specific receptor 4 antagonist inhibits lipopolysaccharide-induced osteoclastogenesis in rat periodontal tissue. J. Periodontol. 2012, 83, 506–513. [Google Scholar] [CrossRef] [PubMed]
- Nishida, E.; Hara, Y.; Kaneko, T.; Ikeda, Y.; Ukai, T.; Kato, I. Bone resorption and local interleukin-1alpha and interleukin-1beta synthesis induced by Actinobacillus actinomycetemcomitans and Porphyromonas gingivalis lipopolysaccharide. J. Periodontal Res. 2001, 36, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Zhang, J.; Fu-Shin, X.Y. Innate immune response of corneal epithelial cells to Staphylococcus aureus infection: Role of peptidoglycan in stimulating proinflammatory cytokine secretion. Investig. Ophthalmol. Vis. Sci. 2004, 45, 3513–3522. [Google Scholar] [CrossRef]
- Hotokezaka, H.; Sakai, E.; Ohara, N.; Hotokezaka, Y.; Gonzales, C.; Matsuo, K.i.; Fujimura, Y.; Yoshida, N.; Nakayama, K. Molecular analysis of RANKL-independent cell fusion of osteoclast-like cells induced by TNF-α, lipopolysaccharide, or peptidoglycan. J. Cell. Biochem. 2007, 101, 122–134. [Google Scholar] [CrossRef]
- Sato, T.; Watanabe, K.; Kumada, H.; Toyama, T.; Tani-Ishii, N.; Hamada, N. Peptidoglycan of Actinomyces naeslundii induces inflammatory cytokine production and stimulates osteoclastogenesis in alveolar bone resorption. Arch. Oral Biol. 2012, 57, 1522–1528. [Google Scholar] [CrossRef]
- Jiang, J.; Zuo, J.; Hurst, I.R.; Holliday, L.S. The synergistic effect of peptidoglycan and lipopolysaccaride on osteoclast formation. Oral Surg. Oral Med. Oral Pathol. Oral Radiol. Endodontol. 2003, 96, 738–743. [Google Scholar] [CrossRef]
- Darveau, R.P. Periodontitis: A polymicrobial disruption of host homeostasis. Nat. Rev. Microbiol. 2010, 8, 481–490. [Google Scholar] [CrossRef]
- Hienz, S.A.; Paliwal, S.; Ivanovski, S. Mechanisms of bone resorption in periodontitis. J. Immunol. Res. 2015, 2015, 615486. [Google Scholar] [CrossRef]
- Hajishengallis, G.; Liang, S.; Payne, M.A.; Hashim, A.; Jotwani, R.; Eskan, M.A.; McIntosh, M.L.; Alsam, A.; Kirkwood, K.L.; Lambris, J.D. Low-abundance biofilm species orchestrates inflammatory periodontal disease through the commensal microbiota and complement. Cell Host Microbe 2011, 10, 497–506. [Google Scholar] [CrossRef]
- Tonetti, M.S.; Henry, G.; Kornman, K.S. Staging and grading of periodontitis: Framework and proposal of a new classification and case definition. J. Periodontol. 2018, 89 (Suppl. 1), S159–S172. [Google Scholar] [CrossRef] [PubMed]
- Lang, N.; Bartokd, P.M.; Cullinan, M.; Jeffcoat, M.; Mombelli, A.; Murakami, S.; Page, R.; Papapanou, P.; Tonetti, M.; Van Dyke, T. Consensus report: Aggressive periodontitis. Ann. Periodontol. 1999, 4, 53. [Google Scholar] [CrossRef]
- Chen, M.X.; Zhong, Y.J.; Dong, Q.Q.; Wong, H.M.; Wen, Y.F. Global, regional, and national burden of severe periodontitis, 1990–2019: An analysis of the Global Burden of Disease Study 2019. J. Clin. Periodontol. 2021, 48, 1165–1188. [Google Scholar] [CrossRef] [PubMed]
- Niccoli, T.; Partridge, L. Ageing as a risk factor for disease. Curr. Biol. 2012, 22, R741–R752. [Google Scholar] [CrossRef] [PubMed]
- Baima, G.; Romandini, M.; Citterio, F.; Romano, F.; Aimetti, M. Periodontitis and accelerated biological aging: A geroscience approach. J. Dent. Res. 2022, 101, 125–132. [Google Scholar] [CrossRef]
- GBD 2017 Oral Disorders Collaborators; Bernabe, E.; Marcenes, W.; Hernandez, C.; Bailey, J.; Abreu, L.; Alipour, V.; Amini, S.; Arabloo, J.; Arefi, Z. Global, regional, and national levels and trends in burden of oral conditions from 1990 to 2017: A systematic analysis for the global burden of disease 2017 study. J. Dent. Res. 2020, 99, 362–373. [Google Scholar] [CrossRef]
- Campisi, J. Aging, cellular senescence, and cancer. Annu. Rev. Physiol. 2013, 75, 685–705. [Google Scholar] [CrossRef]
- D’adda Di Fagagna, F. Living on a break: Cellular senescence as a DNA-damage response. Nat. Rev. Cancer 2008, 8, 512–522. [Google Scholar] [CrossRef]
- Feng, X.; Feng, G.; Xing, J.; Shen, B.; Tan, W.; Huang, D.; Lu, X.; Tao, T.; Zhang, J.; Li, L. Repeated lipopolysaccharide stimulation promotes cellular senescence in human dental pulp stem cells (DPSCs). Cell Tissue Res. 2014, 356, 369–380. [Google Scholar] [CrossRef]
- Toussain, O.; Dumont, P.; Dierick, J.-F.; Pascal, T.; Frippiat, C.; Chainiaux, F.; Sluse, F.; Eliaers, F.; Remacle, J. Stress-induced premature senescence. Essence of life, evolution, stress, and aging. Ann. N. Y. Acad. Sci. 2000, 908, 85–98. [Google Scholar] [CrossRef]
- Kuilman, T.; Michaloglou, C.; Mooi, W.J.; Peeper, D.S. The essence of senescence. Genes Dev. 2010, 24, 2463–2479. [Google Scholar] [CrossRef]
- Waaijer, M.E.; Parish, W.E.; Strongitharm, B.H.; van Heemst, D.; Slagboom, P.E.; de Craen, A.J.; Sedivy, J.M.; Westendorp, R.G.; Gunn, D.A.; Maier, A.B. The number of p16INK4a positive cells in human skin reflects biological age. Aging Cell 2012, 11, 722–725. [Google Scholar] [CrossRef] [PubMed]
- Aquino-Martinez, R.; Eckhardt, B.A.; Rowsey, J.L.; Fraser, D.G.; Khosla, S.; Farr, J.N.; Monroe, D.G. Senescent cells exacerbate chronic inflammation and contribute to periodontal disease progression in old mice. J. Periodontol. 2021, 92, 1483–1495. [Google Scholar] [CrossRef] [PubMed]
- Childs, B.G.; Durik, M.; Baker, D.J.; Van Deursen, J.M. Cellular senescence in aging and age-related disease: From mechanisms to therapy. Nat. Med. 2015, 21, 1424–1435. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Ma, Y.; Zhu, Y.; Zhang, T.; Zhou, Y. Declined expression of histone deacetylase 6 contributes to periodontal ligament stem cell aging. J. Periodontol. 2017, 88, e12–e23. [Google Scholar] [CrossRef]
- Gay, I.C.; Chen, S.; MacDougall, M. Isolation and characterization of multipotent human periodontal ligament stem cells. Orthod. Craniofacial Res. 2007, 10, 149–160. [Google Scholar] [CrossRef]
- Nagatomo, K.; Komaki, M.; Sekiya, I.; Sakaguchi, Y.; Noguchi, K.; Oda, S.; Muneta, T.; Ishikawa, I. Stem cell properties of human periodontal ligament cells. J. Periodontal Res. 2006, 41, 303–310. [Google Scholar] [CrossRef]
- Feng, F.; Akiyama, K.; Liu, Y.; Yamaza, T.; Wang, T.M.; Chen, J.H.; Wang, B.; Huang, G.J.; Wang, S.; Shi, S. Utility of PDL progenitors for in vivo tissue regeneration: A report of 3 cases. Oral Dis. 2010, 16, 20–28. [Google Scholar] [CrossRef]
- Hajishengallis, G. Aging and its impact on innate immunity and inflammation: Implications for periodontitis. J. Oral Biosci. 2014, 56, 30–37. [Google Scholar] [CrossRef]
- Usui, M.; Onizuka, S.; Sato, T.; Kokabu, S.; Ariyoshi, W.; Nakashima, K. Mechanism of alveolar bone destruction in periodontitis—Periodontal bacteria and inflammation. Jpn. Dent. Sci. Rev. 2021, 57, 201–208. [Google Scholar] [CrossRef]
- Socransky, S.; Haffajee, A.; Cugini, M.; Smith, C.; Kent, R., Jr. Microbial complexes in subgingival plaque. J. Clin. Periodontol. 1998, 25, 134–144. [Google Scholar] [CrossRef] [PubMed]
- Sanz, M.; Quirynen, M.; European Workshop in Periodontology Group A. Advances in the aetiology of periodontitis: Group A consensus report of the 5th European Workshop in Periodontology. J. Clin. Periodontol. 2005, 32, 54–56. [Google Scholar] [CrossRef] [PubMed]
- Tatakis, D.N.; Kumar, P.S. Etiology and pathogenesis of periodontal diseases. Dent. Clin. 2005, 49, 491–516. [Google Scholar] [CrossRef] [PubMed]
- Offenbacher, S.; Barros, S.; Singer, R.; Moss, K.; Williams, R.; Beck, J. Periodontal disease at the biofilm–gingival interface. J. Periodontol. 2007, 78, 1911–1925. [Google Scholar] [CrossRef] [PubMed]
- Slots, J. Periodontitis: Facts, fallacies and the future. Periodontology 2000 2017, 75, 7–23. [Google Scholar] [CrossRef] [PubMed]
- Nelson, G.; Wordsworth, J.; Wang, C.; Jurk, D.; Lawless, C.; Martin-Ruiz, C.; von Zglinicki, T. A senescent cell bystander effect: Senescence-induced senescence. Aging Cell 2012, 11, 345–349. [Google Scholar] [CrossRef] [PubMed]
- Acosta, J.C.; Banito, A.; Wuestefeld, T.; Georgilis, A.; Janich, P.; Morton, J.P.; Athineos, D.; Kang, T.-W.; Lasitschka, F.; Andrulis, M. A complex secretory program orchestrated by the inflammasome controls paracrine senescence. Nat. Cell Biol. 2013, 15, 978–990. [Google Scholar] [CrossRef]
- Ohlrich, E.; Cullinan, M.; Seymour, G. The immunopathogenesis of periodontal disease. Aust. Dent. J. 2009, 54, S2–S10. [Google Scholar] [CrossRef]
- Garlet, G.P. Destructive and protective roles of cytokines in periodontitis: A re-appraisal from host defense and tissue destruction viewpoints. J. Dent. Res. 2010, 89, 1349–1363. [Google Scholar] [CrossRef]
- Graves, D. Cytokines that promote periodontal tissue destruction. J. Periodontol. 2008, 79, 1585–1591. [Google Scholar] [CrossRef]
- Madianos, P.; Bobetsis, Y.; Kinane, D. Generation of inflammatory stimuli: How bacteria set up inflammatory responses in the gingiva. J. Clin. Periodontol. 2005, 32, 57–71. [Google Scholar] [CrossRef] [PubMed]
- Genco, C.A.; Potempa, J.; Mikolajczyk-Pawlinska, J.; Travis, J. Role of gingipains R in the pathogenesis of Porphyromonas gingivalis-mediated periodontal disease. Clin. Infect. Dis. 1999, 28, 456–465. [Google Scholar] [CrossRef] [PubMed]
- de Diego, I.; Veillard, F.; Sztukowska, M.N.; Guevara, T.; Potempa, B.; Pomowski, A.; Huntington, J.A.; Potempa, J.; Gomis-Rüth, F.X. Structure and mechanism of cysteine peptidase gingipain K (Kgp), a major virulence factor of Porphyromonas gingivalis in periodontitis. J. Biol. Chem. 2014, 289, 32291–32302. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Nguyen, K.-A.; Potempa, J. Dichotomy of gingipains action as virulence factors: From cleaving substrates with the precision of a surgeon’s knife to a meat chopper-like brutal degradation of proteins. Periodontology 2000 2010, 54, 15. [Google Scholar] [CrossRef] [PubMed]
- Bao, K.; Belibasakis, G.N.; Thurnheer, T.; Aduse-Opoku, J.; Curtis, M.A.; Bostanci, N. Role of Porphyromonas gingivalis gingipains in multi-species biofilm formation. BMC Microbiol. 2014, 14, 258. [Google Scholar] [CrossRef]
- Ito, R.; Ishihara, K.; Shoji, M.; Nakayama, K.; Okuda, K. Hemagglutinin/Adhesin domains of Porphyromonas gingivalis play key roles in coaggregation with Treponema denticola. FEMS Immunol. Med. Microbiol. 2010, 60, 251–260. [Google Scholar] [CrossRef]
- Curtis, M.; Aduse-Opoku, J.; Rangarajan, M. Cysteine proteases of Porphyromonas gingivalis. Crit. Rev. Oral Biol. Med. 2001, 12, 192–216. [Google Scholar] [CrossRef]
- Takeuchi, H.; Sasaki, N.; Yamaga, S.; Kuboniwa, M.; Matsusaki, M.; Amano, A. Porphyromonas gingivalis induces penetration of lipopolysaccharide and peptidoglycan through the gingival epithelium via degradation of junctional adhesion molecule 1. PLoS Pathog. 2019, 15, e1008124. [Google Scholar] [CrossRef]
- Yasuhara, R.; Miyamoto, Y.; Takami, M.; Imamura, T.; Potempa, J.; Yoshimura, K.; Kamijo, R. Lysine-specific gingipain promotes lipopolysaccharide-and active-vitamin D3-induced osteoclast differentiation by degrading osteoprotegerin. Biochem. J. 2009, 419, 159–166. [Google Scholar] [CrossRef]
- Takayanagi, H.; Ogasawara, K.; Hida, S.; Chiba, T.; Murata, S.; Sato, K.; Takaoka, A.; Yokochi, T.; Oda, H.; Tanaka, K. T-cell-mediated regulation of osteoclastogenesis by signalling cross-talk between RANKL and IFN-γ. Nature 2000, 408, 600–605. [Google Scholar] [CrossRef]
- Arron, J.R.; Choi, Y. Bone versus immune system. Nature 2000, 408, 535–536. [Google Scholar] [CrossRef] [PubMed]
- Jiang, C.; Li, Z.; Quan, H.; Xiao, L.; Zhao, J.; Jiang, C.; Wang, Y.; Liu, J.; Gou, Y.; An, S. Osteoimmunology in orthodontic tooth movement. Oral Dis. 2015, 21, 694–704. [Google Scholar] [CrossRef]
- Graves, D.T.; Alshabab, A.; Albiero, M.L.; Mattos, M.; Corrêa, J.D.; Chen, S.; Yang, Y. Osteocytes play an important role in experimental periodontitis in healthy and diabetic mice through expression of RANKL. J. Clin. Periodontol. 2018, 45, 285–292. [Google Scholar] [CrossRef] [PubMed]
- Thorbert-Mros, S.; Larsson, L.; Berglundh, T. Cellular composition of long-standing gingivitis and periodontitis lesions. J. Periodontal Res. 2015, 50, 535–543. [Google Scholar] [CrossRef] [PubMed]
- Schulze-Späte, U.; Turner, R.; Wang, Y.; Chao, R.; Schulze, P.C.; Phipps, K.; Orwoll, E.; Dam, T.-T. Relationship of bone metabolism biomarkers and periodontal disease: The osteoporotic fractures in men (MrOS) study. J. Clin. Endocrinol. Metab. 2015, 100, 2425–2433. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Cytokines/Chemokines/ Cellular Signalling Pathways | Immunosenescence | Age-Related Chronic Inflammation |
|---|---|---|
| IL-1α | ↑↑↑ | ↑ |
| IL-1β | ↑↑ | ↑↑ |
| IL-6 | ↑↑↑ | ↑ |
| TNF-α | - | ↑ |
| IL-8 | ↑↑↑ | ↑ |
| CXCL10 | ↑↑↑ | - |
| IL-12 | ↑ | ↑↑ |
| IFN-α | - | ↑ |
| iNOS | - | ↑↑↑ |
| GSK3-β | ↑ | ↑ |
| MAPK | ↑ | ↑ |
| PKB | ↑ | ↑↑↑ |
| NF-κB | ↑ | ↑↑↑ |
| References | [19,21,22,23] | [24,25,26,27] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhu, L.; Tang, Z.; Hu, R.; Gu, M.; Yang, Y. Ageing and Inflammation: What Happens in Periodontium? Bioengineering 2023, 10, 1274. https://doi.org/10.3390/bioengineering10111274
Zhu L, Tang Z, Hu R, Gu M, Yang Y. Ageing and Inflammation: What Happens in Periodontium? Bioengineering. 2023; 10(11):1274. https://doi.org/10.3390/bioengineering10111274
Chicago/Turabian StyleZhu, Luying, Zhongyuan Tang, Renjie Hu, Min Gu, and Yanqi Yang. 2023. "Ageing and Inflammation: What Happens in Periodontium?" Bioengineering 10, no. 11: 1274. https://doi.org/10.3390/bioengineering10111274