
Investigation on UV Degradation and Mechanism of 6:2 Fluorotelomer Sulfonamide Alkyl Betaine, Based on Model Compound Perfluorooctanoic Acid

 , , and
, , and
Abstract
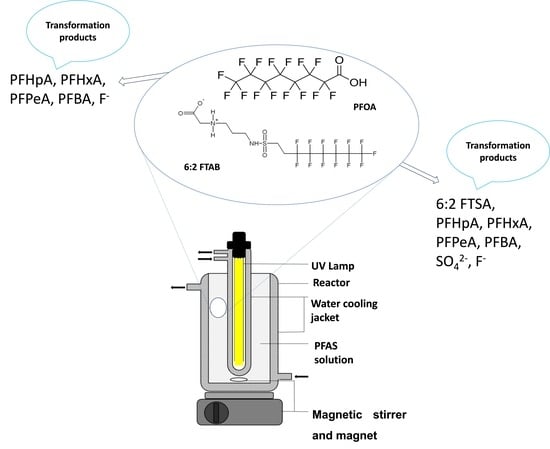
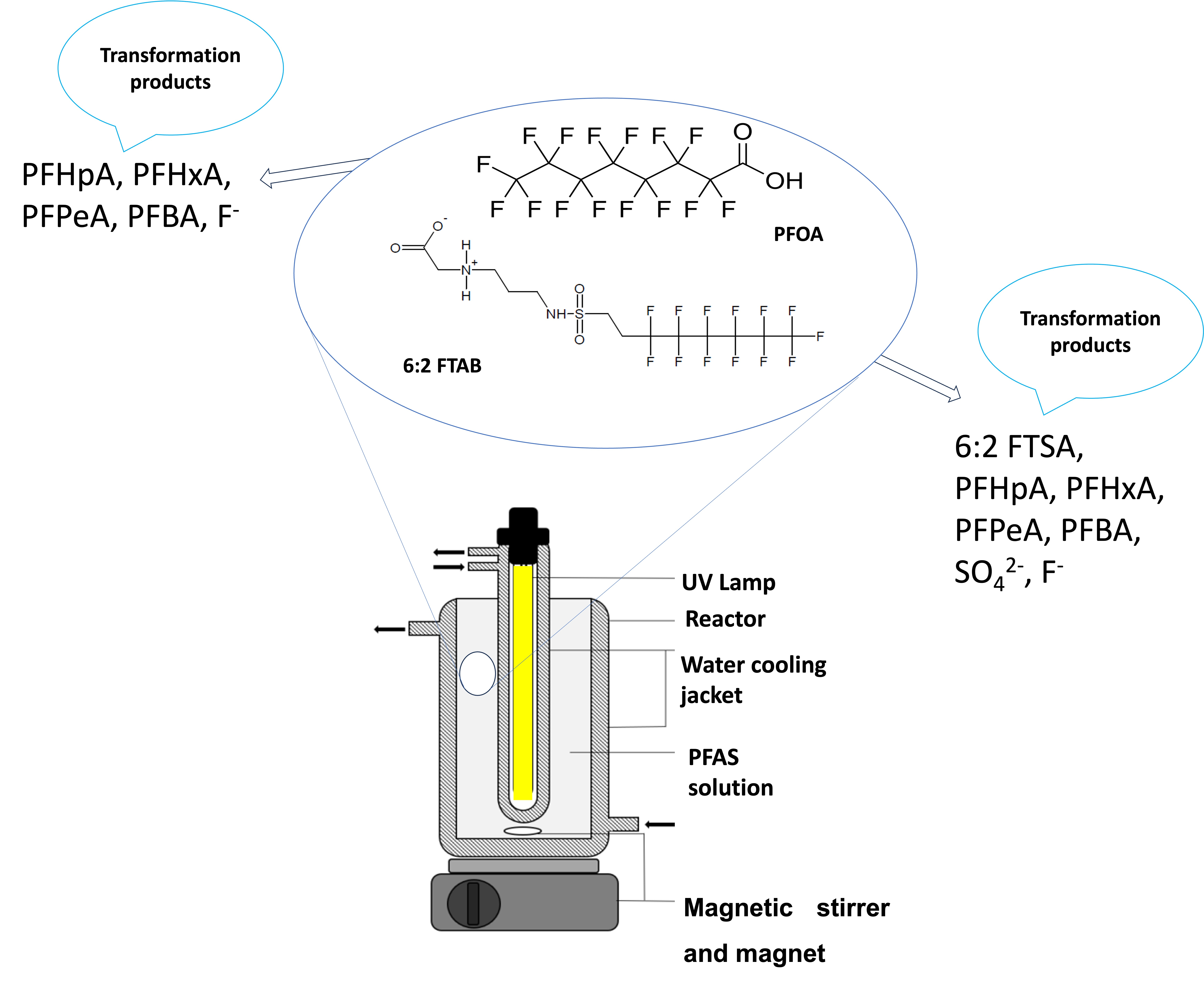
:
1. Introduction
2. Materials and Methods
2.1. Chemicals
2.2. Experimental Set-Up and Photolysis of PFOA and 6:2 FTAB
2.3. Radical Scavenging Experiments
2.4. Quantification of PFOA, 6:2 FTAB, and Their Metabolites by LC-MS
2.5. Fluoride and Sulfate Measurement by Ion Chromatography
2.6. Dissolved Oxygen and Hydrogen Peroxide Measurements
3. Results and Discussion
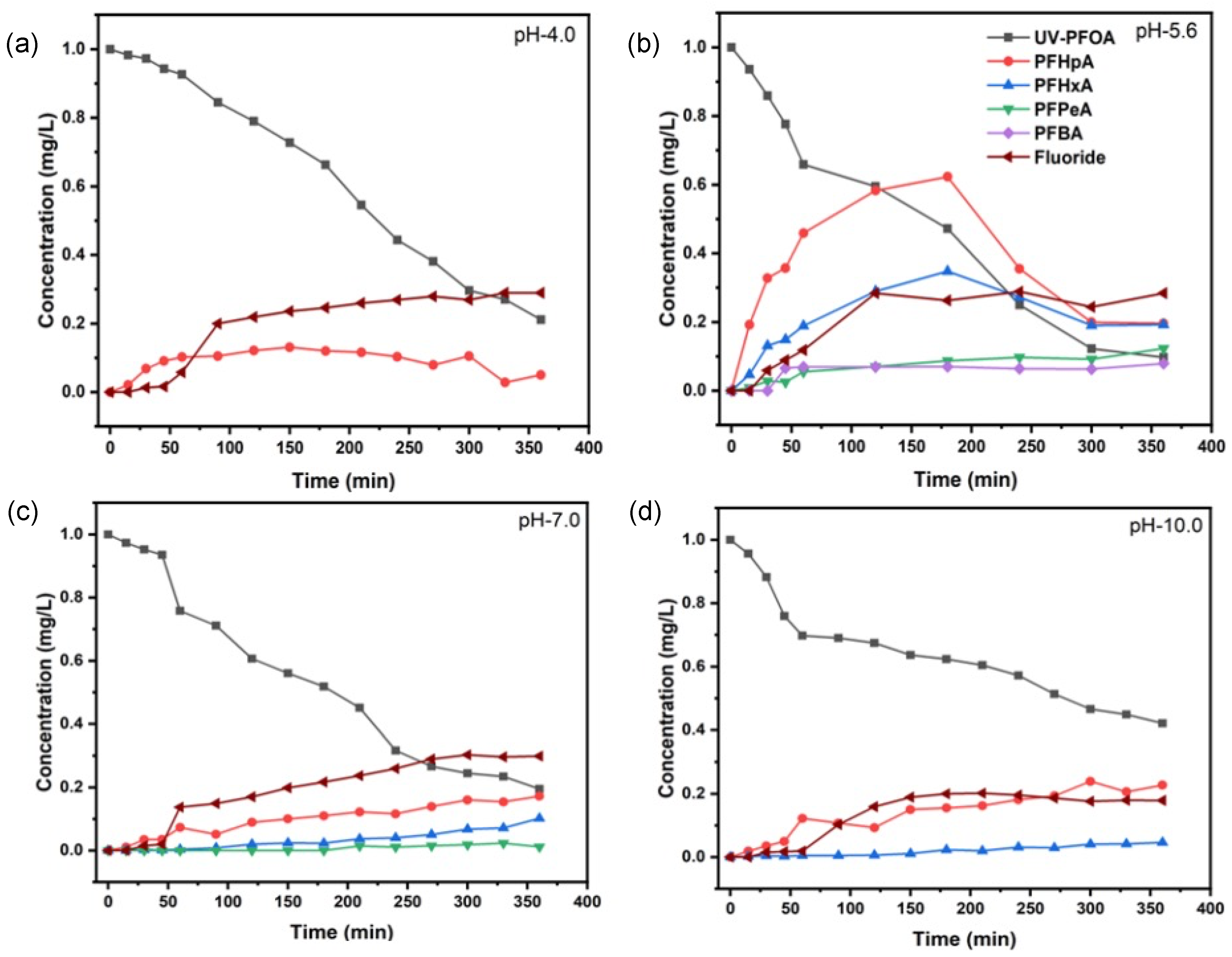
3.1. Decomposition of PFOA at Different pH Values
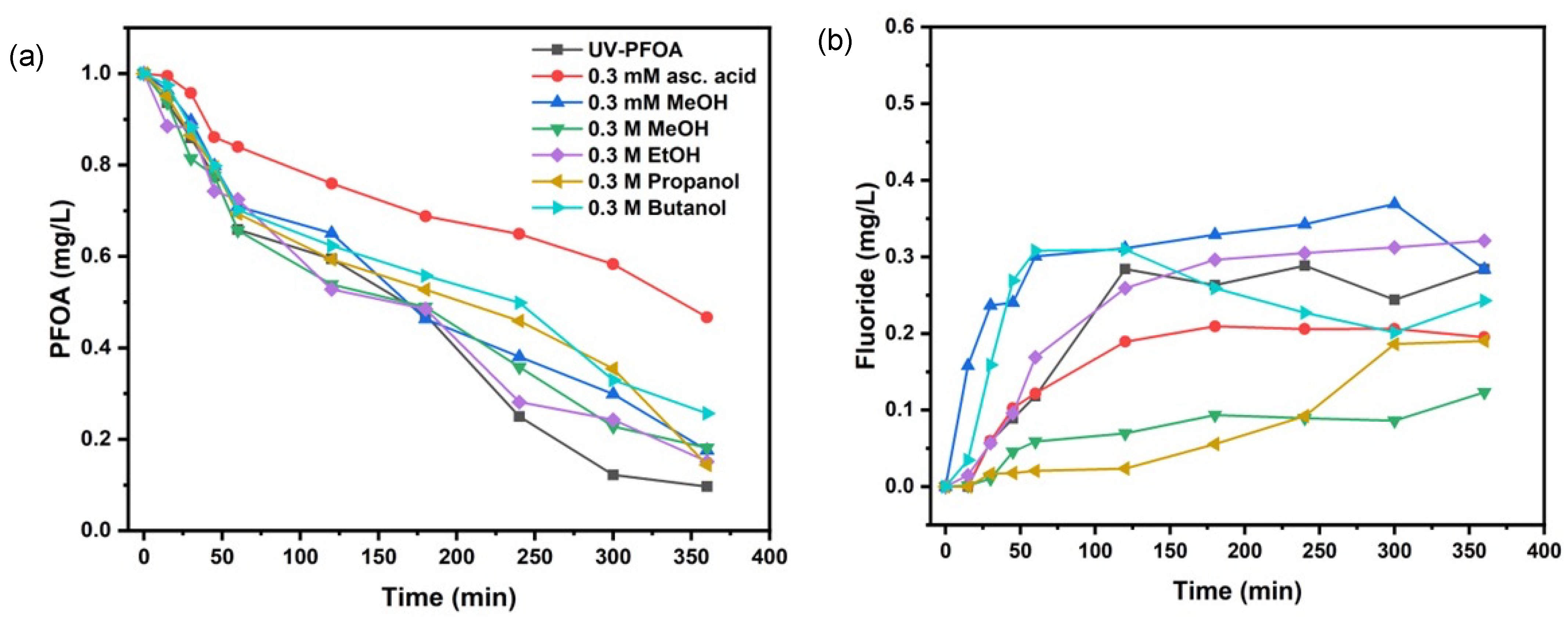
3.2. Scavengers’ Experiments for PFOA Decomposition
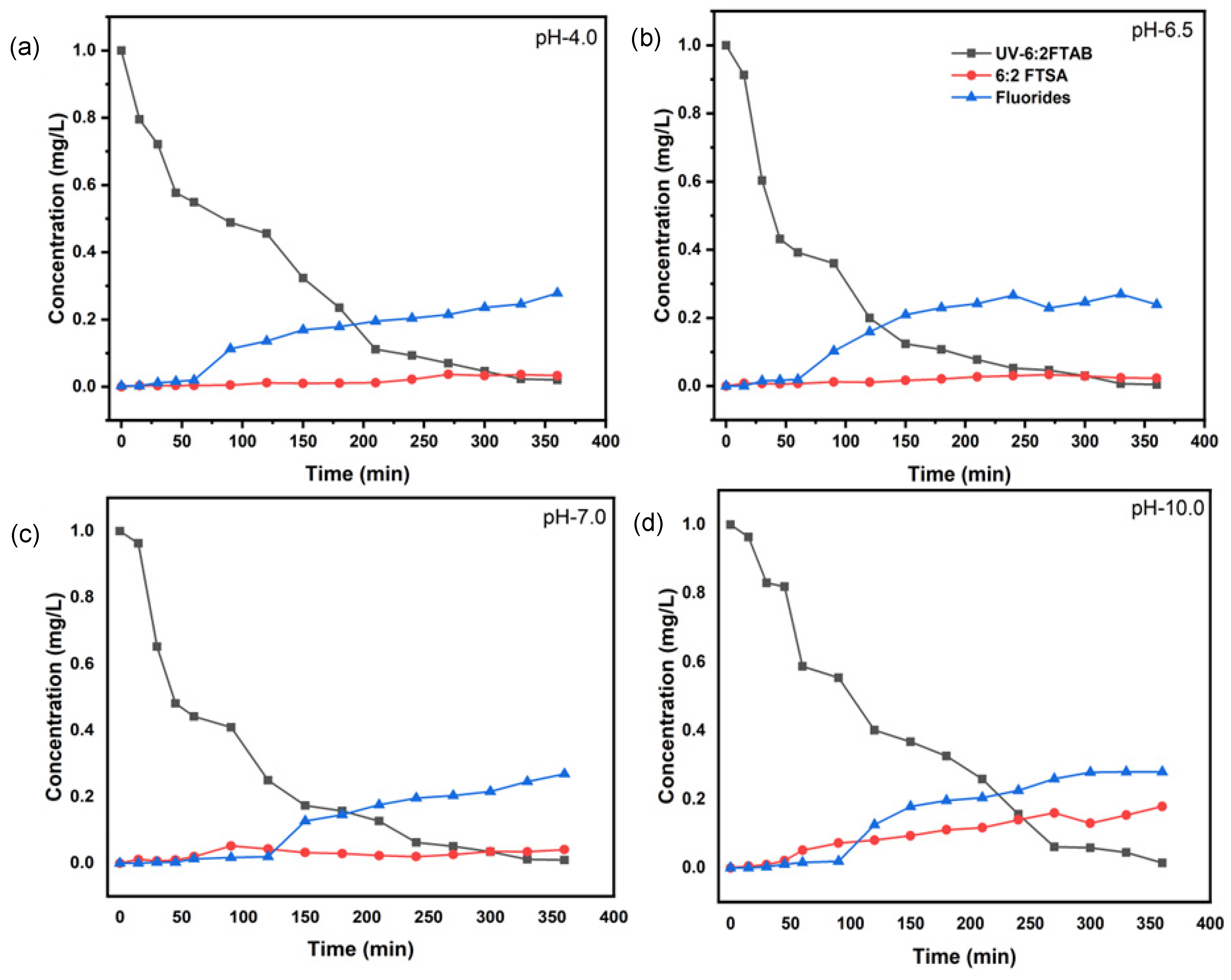
3.3. Decomposition of 6:2 FTAB at Different pH Values
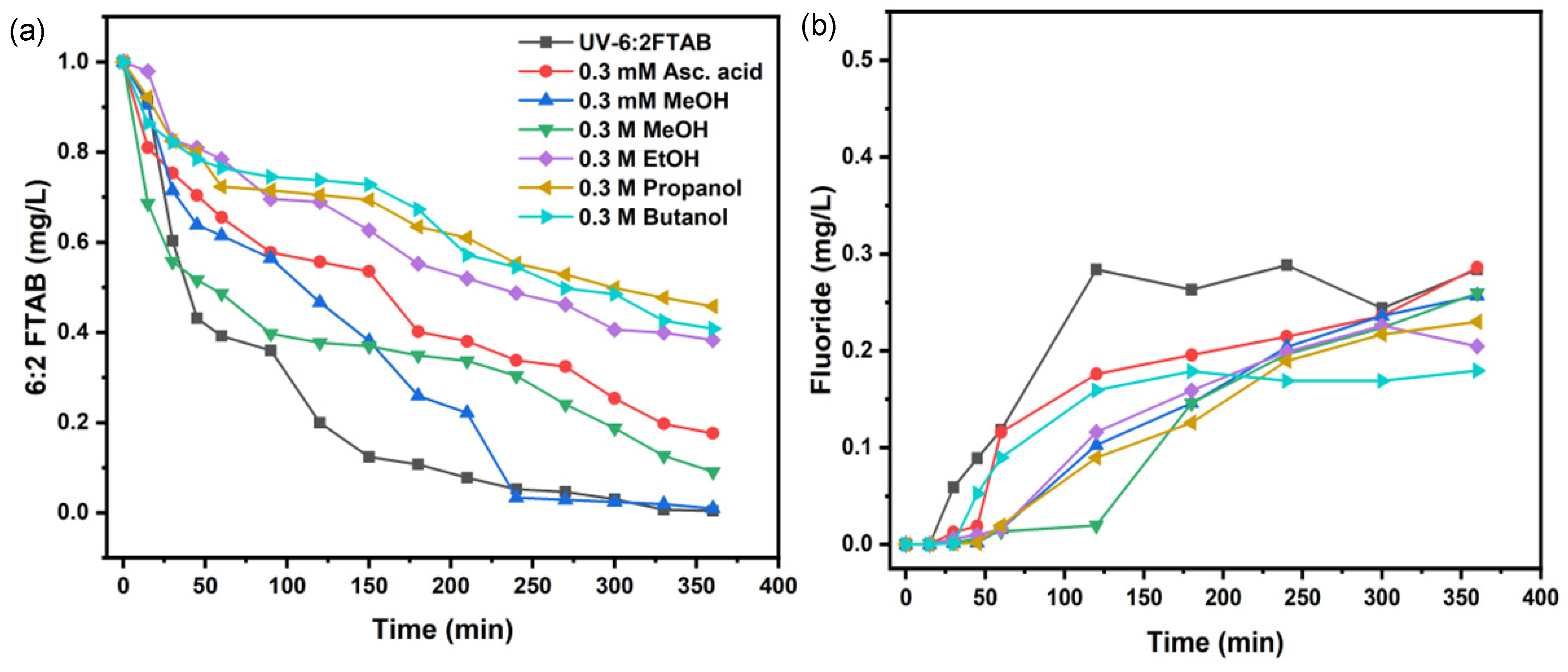
3.4. Scavengers Experiments for 6:2 FTAB Decomposition
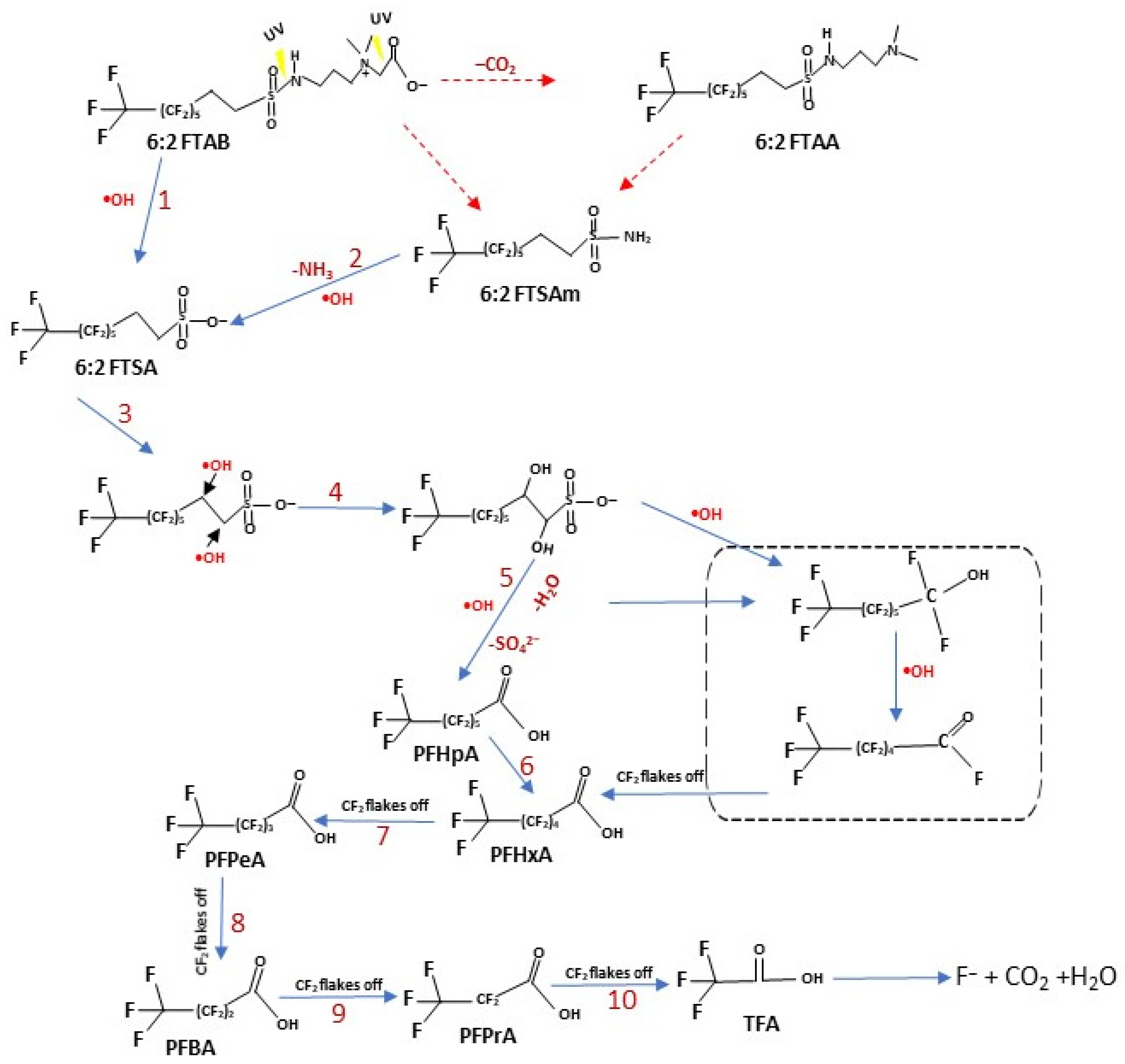
3.5. Proposed Mechanism of 6:2 FTAB Decomposition
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cousins, I.T.; DeWitt, J.C.; Glüge, J.; Goldenman, G.; Herzke, D.; Lohmann, R.; Ng, C.A.; Scheringer, M.; Wang, Z. The High Persistence of PFAS Is Sufficient for Their Management as a Chemical Class. Environ. Sci. Process. Impacts 2020, 22, 2307–2312. [Google Scholar] [CrossRef]
- Wang, Z.; DeWitt, J.C.; Higgins, C.P.; Cousins, I.T. A Never-Ending Story of per-and Polyfluoroalkyl Substances (PFASs)? Environ. Sci. Technol. 2017, 51, 2508–2518. [Google Scholar] [CrossRef]
- Wang, T.; Wang, P.; Meng, J.; Liu, S.; Lu, Y.; Khim, J.S.; Giesy, J.P. A Review of Sources, Multimedia Distribution and Health Risks of Perfluoroalkyl Acids (PFAAs) in China. Chemosphere 2015, 129, 87–99. [Google Scholar] [CrossRef]
- Kotthoff, M.; Müller, J.; Jürling, H.; Schlummer, M.; Fiedler, D. Perfluoroalkyl and Polyfluoroalkyl Substances in Consumer Products. Environ. Sci. Pollut. Res. 2015, 22, 14546–14559. [Google Scholar] [CrossRef]
- Guo, Z.; Liu, X.; Krebs, K.A.; Roache, N.F. Perfluorocarboxylic Acid Content in 116 Articles of Commerce; U.S. Environmental Protection Agency, Office of Research and Development: Research Triangle Park, NC, USA, 2009.
- Xin, X.; Kim, J.; Ashley, D.C.; Huang, C.-H. Degradation and Defluorination of Per-and Polyfluoroalkyl Substances by Direct Photolysis at 222 Nm. ACS Est Water 2023, 3, 2776–2785. [Google Scholar] [CrossRef]
- Leung, S.C.E.; Shukla, P.; Chen, D.; Eftekhari, E.; An, H.; Zare, F.; Ghasemi, N.; Zhang, D.; Nguyen, N.-T.; Li, Q. Emerging Technologies for PFOS/PFOA Degradation and Removal: A Review. Sci. Total Environ. 2022, 827, 153669. [Google Scholar] [CrossRef]
- Tahziz, A.; Ruzi, I.I.; Yahaya, N.; Mohamed, R.; Ishak, A.R.; Edinur, H.A.; Aziz, M.Y. Occurrence and Toxicology Aspects of Perfluorooctane Sulfonate (PFOS) and Perfluorooctanoic Acid (PFOA) in the Environment and Food: A Review. Malays. J. Med. Heal. Sci. 2021, 17, 165–175. [Google Scholar]
- Shi, G.; Xie, Y.; Guo, Y.; Dai, J. 6:2 Fluorotelomer Sulfonamide Alkylbetaine (6:2 FTAB), a Novel Perfluorooctane Sulfonate Alternative, Induced Developmental Toxicity in Zebrafish Embryos. Aquat. Toxicol. 2018, 195, 24–32. [Google Scholar] [CrossRef]
- De Solla, S.R.; De Silva, A.O.; Letcher, R.J. Highly Elevated Levels of Perfluorooctane Sulfonate and Other Perfluorinated Acids Found in Biota and Surface Water Downstream of an International Airport, Hamilton, Ontario, Canada. Environ. Int. 2012, 39, 19–26. [Google Scholar] [CrossRef]
- Schultz, M.M.; Barofsky, D.F.; Field, J.A. Quantitative Determination of Fluorotelomer Sulfonates in Groundwater by LC MS/MS. Environ. Sci. Technol. 2004, 38, 1828–1835. [Google Scholar] [CrossRef]
- Houtz, E.F.; Higgins, C.P.; Field, J.A.; Sedlak, D.L. Persistence of Perfluoroalkyl Acid Precursors in AFFF-Impacted Groundwater and Soil. Environ. Sci. Technol. 2013, 47, 8187–8195. [Google Scholar] [CrossRef]
- Houtz, E.F.; Sutton, R.; Park, J.-S.; Sedlak, M. Poly-and Perfluoroalkyl Substances in Wastewater: Significance of Unknown Precursors, Manufacturing Shifts, and Likely AFFF Impacts. Water Res. 2016, 95, 142–149. [Google Scholar] [CrossRef]
- Mazumder, N.-U.-S.; Hossain, M.T.; Jahura, F.T.; Girase, A.; Hall, A.S.; Lu, J.; Ormond, R.B. Firefighters’ Exposure to per-and Polyfluoroalkyl Substances (PFAS) as an Occupational Hazard: A Review. Front. Mater. 2023, 10, 1143411. [Google Scholar] [CrossRef] [PubMed]
- Butt, C.M.; Muir, D.C.G.; Mabury, S.A. Biotransformation Pathways of Fluorotelomer-based Polyfluoroalkyl Substances: A Review. Environ. Toxicol. Chem. 2014, 33, 243–267. [Google Scholar] [CrossRef]
- Dinglasan, M.J.A.; Ye, Y.; Edwards, E.A.; Mabury, S.A. Fluorotelomer Alcohol Biodegradation Yields Poly-and Perfluorinated Acids. Environ. Sci. Technol. 2004, 38, 2857–2864. [Google Scholar] [CrossRef] [PubMed]
- Harding-Marjanovic, K.C.; Houtz, E.F.; Yi, S.; Field, J.A.; Sedlak, D.L.; Alvarez-Cohen, L. Aerobic Biotransformation of Fluorotelomer Thioether Amido Sulfonate (Lodyne) in AFFF-Amended Microcosms. Environ. Sci. Technol. 2015, 49, 7666–7674. [Google Scholar] [CrossRef]
- Weiner, B.; Yeung, L.W.Y.; Marchington, E.B.; D’Agostino, L.A.; Mabury, S.A. Organic Fluorine Content in Aqueous Film Forming Foams (AFFFs) and Biodegradation of the Foam Component 6: 2 Fluorotelomermercaptoalkylamido Sulfonate (6: 2 FTSAS). Environ. Chem. 2013, 10, 486–493. [Google Scholar] [CrossRef]
- Gauthier, S.A.; Mabury, S.A. Aqueous Photolysis of 8: 2 Fluorotelomer Alcohol. Environ. Toxicol. Chem. An Int. J. 2005, 24, 1837–1846. [Google Scholar] [CrossRef]
- Mojiri, A.; Zhou, J.L.; Ozaki, N.; KarimiDermani, B.; Razmi, E.; Kasmuri, N. Occurrence of Per-and Polyfluoroalkyl Substances in Aquatic Environments and Their Removal by Advanced Oxidation Processes. Chemosphere 2023, 330, 138666. [Google Scholar] [CrossRef]
- Jing, C.; Zhang, P.; Jian, L.I.U. Photodegradation of Perfluorooctanoic Acid by 185 Nm Vacuum Ultraviolet Light. J. Environ. Sci. 2007, 19, 387–390. [Google Scholar]
- Bai, L.; Jiang, Y.; Xia, D.; Wei, Z.; Spinney, R.; Dionysiou, D.D.; Minakata, D.; Xiao, R.; Xie, H.-B.; Chai, L. Mechanistic Understanding of Superoxide Radical-Mediated Degradation of Perfluorocarboxylic Acids. Environ. Sci. Technol. 2021, 56, 624–633. [Google Scholar] [CrossRef]
- Chen, M.-J.; Lo, S.-L.; Lee, Y.-C.; Kuo, J.; Wu, C.-H. Decomposition of Perfluorooctanoic Acid by Ultraviolet Light Irradiation with Pb-Modified Titanium Dioxide. J. Hazard. Mater. 2016, 303, 111–118. [Google Scholar] [CrossRef]
- Javed, H.; Lyu, C.; Sun, R.; Zhang, D.; Alvarez, P.J.J. Discerning the Inefficacy of Hydroxyl Radicals during Perfluorooctanoic Acid Degradation. Chemosphere 2020, 247, 125883. [Google Scholar] [CrossRef]
- Gomez-Ruiz, B.; Ribao, P.; Diban, N.; Rivero, M.J.; Ortiz, I.; Urtiaga, A. Photocatalytic Degradation and Mineralization of Perfluorooctanoic Acid (PFOA) Using a Composite TiO2− RGO Catalyst. J. Hazard. Mater. 2018, 344, 950–957. [Google Scholar] [CrossRef] [PubMed]
- Da Silva-Rackov, C.K.O.; Lawal, W.A.; Nfodzo, P.A.; Vianna, M.M.G.R.; do Nascimento, C.A.O.; Choi, H. Degradation of PFOA by Hydrogen Peroxide and Persulfate Activated by Iron-Modified Diatomite. Appl. Catal. B Environ. 2016, 192, 253–259. [Google Scholar] [CrossRef]
- Metz, J.; Javed, H.; Mathieu, J.; Long, M.; Alvarez, P.J.J. Comment on “Mechanistic Understanding of Superoxide Radical-Mediated Degradation of Perfluorocarboxylic Acids”. Environ. Sci. Technol. 2022, 56, 5287–5288. [Google Scholar] [CrossRef]
- Javed, H.; Metz, J.; Eraslan, T.C.; Mathieu, J.; Wang, B.; Wu, G.; Tsai, A.-L.; Wong, M.S.; Alvarez, P.J.J. Discerning the Relevance of Superoxide in PFOA Degradation. Environ. Sci. Technol. Lett. 2020, 7, 653–658. [Google Scholar] [CrossRef]
- Chowdhury, N.; Choi, H. Photocatalytic Degradation of Perfluorooctanoic Acid on Pb-doped TiO2 Coated with Reduced Graphene Oxide. Water Environ. Res. 2023, 95, e10871. [Google Scholar] [CrossRef] [PubMed]
- Hori, H.; Hayakawa, E.; Einaga, H.; Kutsuna, S.; Koike, K.; Ibusuki, T.; Kiatagawa, H.; Arakawa, R. Decomposition of Environmentally Persistent Perfluorooctanoic Acid in Water by Photochemical Approaches. Environ. Sci. Technol. 2004, 38, 6118–6124. [Google Scholar] [CrossRef] [PubMed]
- Deng, Y.; Liang, Z.; Lu, X.; Chen, D.; Li, Z.; Wang, F. The Degradation Mechanisms of Perfluorooctanoic Acid (PFOA) and Perfluorooctane Sulfonic Acid (PFOS) by Different Chemical Methods: A Critical Review. Chemosphere 2021, 283, 131168. [Google Scholar] [CrossRef]
- Chen, M.-J.; Lo, S.-L.; Lee, Y.-C.; Huang, C.-C. Photocatalytic Decomposition of Perfluorooctanoic Acid by Transition-Metal Modified Titanium Dioxide. J. Hazard. Mater. 2015, 288, 168–175. [Google Scholar] [CrossRef]
- Chen, Y.-C.; Lo, S.-L.; Kuo, J. Effects of Titanate Nanotubes Synthesized by a Microwave Hydrothermal Method on Photocatalytic Decomposition of Perfluorooctanoic Acid. Water Res. 2011, 45, 4131–4140. [Google Scholar] [CrossRef]
- Huang, J.; Wang, X.; Pan, Z.; Li, X.; Ling, Y.; Li, L. Efficient Degradation of Perfluorooctanoic Acid (PFOA) by Photocatalytic Ozonation. Chem. Eng. J. 2016, 296, 329–334. [Google Scholar] [CrossRef]
- Lin, H.; Niu, J.; Ding, S.; Zhang, L. Electrochemical Degradation of Perfluorooctanoic Acid (PFOA) by Ti/SnO2–Sb, Ti/SnO2–Sb/PbO2 and Ti/SnO2–Sb/MnO2 Anodes. Water Res. 2012, 46, 2281–2289. [Google Scholar] [CrossRef] [PubMed]
- Cao, L.; Ma, D.; Zhou, Z.; Xu, C.; Cao, C.; Zhao, P.; Huang, Q. Efficient Photocatalytic Degradation of Herbicide Glyphosate in Water by Magnetically Separable and Recyclable BiOBr/Fe3O4 Nanocomposites under Visible Light Irradiation. Chem. Eng. J. 2019, 368, 212–222. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, P. Effects of PH on Photochemical Decomposition of Perfluorooctanoic Acid in Different Atmospheres by 185 Nm Vacuum Ultraviolet. J. Environ. Sci. 2014, 26, 2207–2214. [Google Scholar] [CrossRef] [PubMed]
- Qu, Y.; Zhang, C.-J.; Chen, P.; Zhou, Q.; Zhang, W.-X. Effect of Initial Solution PH on Photo-Induced Reductive Decomposition of Perfluorooctanoic Acid. Chemosphere 2014, 107, 218–223. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Teng, Y.; Wang, W.; Hong, R.; Huang, L.; Wang, X.; Zhu, F.; Li, H.; Hao, S.; Wu, B.; et al. Enhanced UV Photoreductive Destruction of Perfluorooctanoic Acid in the Presence of Alcohols: Synergistic Mechanism of Hydroxyl Radical Quenching and Solvent Effect. Appl. Catal. B Environ. 2022, 316, 121652. [Google Scholar] [CrossRef]
- Gonzalez, M.G.; Oliveros, E.; Wörner, M.; Braun, A.M. Vacuum-Ultraviolet Photolysis of Aqueous Reaction Systems. J. Photochem. Photobiol. C Photochem. Rev. 2004, 5, 225–246. [Google Scholar] [CrossRef]
- Giri, R.R.; Ozaki, H.; Okada, T.; Taniguchi, S.; Takanami, R. Factors Influencing UV Photodecomposition of Perfluorooctanoic Acid in Water. Chem. Eng. J. 2012, 180, 197–203. [Google Scholar] [CrossRef]
- Jin, L.; Zhang, P. Photochemical Decomposition of Perfluorooctane Sulfonate (PFOS) in an Anoxic Alkaline Solution by 185 Nm Vacuum Ultraviolet. Chem. Eng. J. 2015, 280, 241–247. [Google Scholar] [CrossRef]
- Chen, J.; Zhang, P. Photodegradation of Perfluorooctanoic Acid in Water under Irradiation of 254 Nm and 185 Nm Light by Use of Persulfate. Water Sci. Technol. 2006, 54, 317–325. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Junker, A.L.; Wen, J.; Ahrens, L.; Sillanpää, M.; Tian, J.; Cui, F.; Vergeynst, L.; Wei, Z. A Recent Overview of Per- and Polyfluoroalkyl Substances (PFAS) Removal by Functional Framework Materials. Chem. Eng. J. 2023, 452, 139202. [Google Scholar] [CrossRef]
- Karbassiyazdi, E.; Kasula, M.; Modak, S.; Pala, J.; Kalantari, M.; Altaee, A.; Esfahani, M.R.; Razmjou, A. A Juxtaposed Review on Adsorptive Removal of PFAS by Metal-Organic Frameworks (MOFs) with Carbon-Based Materials, Ion Exchange Resins, and Polymer Adsorbents. Chemosphere 2023, 311, 136933. [Google Scholar] [CrossRef] [PubMed]
- Juve, J.-M.A.; Donoso Reece, J.A.; Wong, M.S.; Wei, Z.; Ateia, M. Photocatalysts for Chemical-Free PFOA Degradation—What We Know and Where We Go from Here? J. Hazard. Mater. 2024, 462, 132651. [Google Scholar] [CrossRef] [PubMed]
- Hori, H.; Yamamoto, A.; Koike, K.; Kutsuna, S.; Osaka, I.; Arakawa, R. Photochemical Decomposition of Environmentally Persistent Short-Chain Perfluorocarboxylic Acids in Water Mediated by Iron (II)/(III) Redox Reactions. Chemosphere 2007, 68, 572–578. [Google Scholar] [CrossRef]
- Kuhn, R.; Bryant, I.M.; Jensch, R.; Liebsch, S.; Martienssen, M. Photolysis of Hexamethylenediaminetetra (Methylenephosphonic Acid)(HDTMP) Using Manganese and Hydrogen Peroxide. Emerg. Contam. 2020, 6, 10–19. [Google Scholar] [CrossRef]
- Ahmed, N.; Vione, D.; Rivoira, L.; Carena, L.; Castiglioni, M.; Bruzzoniti, M.C. A Review on the Degradation of Pollutants by Fenton-Like Systems Based on Zero-Valent Iron and Persulfate: Effects of Reduction Potentials, PH, and Anions Occurring in Waste Waters. Molecules 2021, 26, 4584. [Google Scholar] [CrossRef] [PubMed]
- Cheng, M.; Zeng, G.; Huang, D.; Lai, C.; Xu, P.; Zhang, C.; Liu, Y. Hydroxyl Radicals Based Advanced Oxidation Processes (AOPs) for Remediation of Soils Contaminated with Organic Compounds: A Review. Chem. Eng. J. 2016, 284, 582–598. [Google Scholar] [CrossRef]
- Kuhn, R.; Jensch, R.; Bryant, I.M.; Fischer, T.; Liebsch, S.; Martienssen, M. Photodegradation of Ethylenediaminetetra (Methylenephosphonic Acid)–The Effect of the System Configuration. J. Photochem. Photobiol. A Chem. 2020, 388, 112192. [Google Scholar] [CrossRef]
- Wang, L.; Li, B.; Dionysiou, D.D.; Chen, B.; Yang, J.; Li, J. Overlooked Formation of H2O2 during the Hydroxyl Radical-Scavenging Process When Using Alcohols as Scavengers. Environ. Sci. Technol. 2022, 56, 3386–3396. [Google Scholar] [CrossRef] [PubMed]
- Dewhirst, R.A.; Fry, S.C. The Oxidation of Dehydroascorbic Acid and 2, 3-Diketogulonate by Distinct Reactive Oxygen Species. Biochem. J. 2018, 475, 3451–3470. [Google Scholar] [CrossRef] [PubMed]
- Poljsak, B.; Ionescu, J.G. Pro-Oxidant vs. Antioxidant Effects of Vitamin C. In Handbook of Vitamin C Research: Daily Requirements, Dietary Sources and Adverse Effects; Nova Biomedical Books: Hauppauge, NY, USA, 2009; p. 153. [Google Scholar]
- Plumlee, M.H.; McNeill, K.; Reinhard, M. Indirect Photolysis of Perfluorochemicals: Hydroxyl Radical-Initiated Oxidation of N-Ethyl Perfluorooctane Sulfonamido Acetate (N-EtFOSAA) and Other Perfluoroalkanesulfonamides. Environ. Sci. Technol. 2009, 43, 3662–3668. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Huang, J.; Zhang, K.; Yu, G.; Deng, S.; Wang, B. Stability of 6:2 Fluorotelomer Sulfonate in Advanced Oxidation Processes: Degradation Kinetics and Pathway. Environ. Sci. Pollut. Res. 2014, 21, 4634–4642. [Google Scholar] [CrossRef] [PubMed]
- D’Agostino, L.A.; Mabury, S.A. Identification of Novel Fluorinated Surfactants in Aqueous Film Forming Foams and Commercial Surfactant Concentrates. Environ. Sci. Technol. 2014, 48, 121–129. [Google Scholar] [CrossRef]
- Trouborst, L. Aqueous Photolysis of 6:2 Fluorotelomer Sulfonamide Alkylbetaine. Master’s Thesis, University of Toronto, Toronto, ON, Canada, 2016. [Google Scholar]
- Martin, J.W.; Mabury, S.A.; O’Brien, P.J. Metabolic Products and Pathways of Fluorotelomer Alcohols in Isolated Rat Hepatocytes. Chem. Biol. Interact. 2005, 155, 165–180. [Google Scholar] [CrossRef]
- Esfahani, E.B.; Zeidabadi, F.A.; Zhang, S.; Mohseni, M. Photo-Chemical/Catalytic Oxidative/Reductive Decomposition of per-and Poly-Fluoroalkyl Substances (PFAS), Decomposition Mechanisms and Effects of Key Factors: A Review. Environ. Sci. Water Res. Technol. 2022, 8, 698–728. [Google Scholar] [CrossRef]
- Marchington, E.B. Identification of Known and Novel Fluorinated Compounds in AFFF Via£ ʹ9-NMR, LC-MS/MS, and LC-Quad-TOFMS, and the Aerobic Biodegradation of 6: 2 FtS. Ph.D. Thesis, University of Toronto, Toronto, ON, Canada, 2009. [Google Scholar]
- D’Agostino, L.A. Environmental Chemistry of Perfluoroalkyl and Polyfluoroalkyl Substances in Aqueous Film Forming Foams. Ph.D. Thesis, University of Toronto, Toronto, ON, Canada, 2017. [Google Scholar]
- Gomez-Ruiz, B.; Gómez-Lavín, S.; Diban, N.; Boiteux, V.; Colin, A.; Dauchy, X.; Urtiaga, A. Boron Doped Diamond Electrooxidation of 6: 2 Fluorotelomers and Perfluorocarboxylic Acids. Application to Industrial Wastewaters Treatment. J. Electroanal. Chem. 2017, 798, 51–57. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| pH Value | k (s−1) | t1/2 (min) | Decomposition (%) | Fluoride Release (%) |
|---|---|---|---|---|
| 4.0 | (8.85 ± 1.13) × 10−5 | 130 ± 2 | 85.2 ± 1.6 | 29.0 ± 3.5 |
| 5.6 | (1.08 ± 0.30) × 10−4 | 107 ± 2 | 90.3 ± 2.5 | 28.8 ± 4.8 |
| 7.0 | (7.57 ± 1.70) × 10−5 | 152 ± 3 | 80.5 ± 4.0 | 30.3 ± 5.4 |
| 10.0 | (4.00 ± 1.15) × 10−5 | 288 ± 3 | 57.9 ± 1.8 | 20.2 ± 3.5 |
| Scavenger & Initial pH | k (s−1) | t1/2 (min) | Decomposition (%) | Fluoride Release (%) |
|---|---|---|---|---|
| 0.3 mM Methanol—pH-5.1 | (8.06 ± 1.5) × 10−5 | 143 ± 3 | 82.6 ±2.9 | 36.9 ± 1.3 |
| 0.3 M Methanol—pH-5.1 | (7.91 ± 0.2) × 10−5 | 146 ± 4 | 81.9 ± 2.7 | 12.3 ± 4.3 |
| 0.3 M Ethanol—pH-5.7 | (8.74 ± 1.2) × 10−5 | 132 ± 5 | 84.9 ± 1.9 | 32.1 ± 3.2 |
| 0.3 M Propanol—pH-5.8 | (9.00 ± 1.9) × 10−5 | 128 ± 5 | 85.7 ± 1.5 | 19.0 ± 2.8 |
| 0.3 M Butanol—pH-5.7 | (6.30 ± 1.5) × 10−5 | 183 ± 3 | 74.3 ± 4.2 | 30.8 ± 4.6 |
| 0.3 mM Ascorbic acid *—pH-4.3 | (3.52 ± 1.3) × 10−5 | 327 ± 6 | 53.3 ± 5.2 | 20.9 ± 4.2 |
| pH Value | k (s−1) | t1/2 (min) | 6:2 FTAB Decomposition (%) | Fluoride Release (%) |
|---|---|---|---|---|
| 4.0 | (1.80 ± 0.06) × 10−4 | 64.1 ± 7.2 | 98.0 ± 3.1 | 27.9 ± 5.0 |
| 6.5 | (2.52 ± 0.16) × 10−4 | 45.7 ± 1.7 | 99.6 ± 0.5 | 27.0 ± 4.7 |
| 7.0 | (2.16 ± 0.29) × 10−4 | 53.3 ± 3.9 | 99.1 ± 0.2 | 26.9 ± 3.2 |
| 10.0 | (1.97 ± 0.14) × 10−4 | 58.4 ± 6.2 | 98.6 ± 1.4 | 28.0 ± 4.2 |
| Scavengers | k(s−1) | t1/2 (min) | 6:2 FTAB Decomposition (%) | Fluoride Release (%) |
|---|---|---|---|---|
| 0.3 mM Methanol | (2.15 ± 0.12) × 10−4 | 54 ± 4 | 99.0 ± 0.7 | 25.7 ± 4.4 |
| 0.3 M Methanol | (1.10 ± 0.24) × 10−4 | 104 ± 8 | 90.9 ± 4.3 | 26.0 ± 2.6 |
| 0.3 M Ethanol | (4.44 ± 0.41) × 10−5 | 260 ± 7 | 61.7 ± 2.9 | 22.6 ± 3.7 |
| 0.3 M Propanol | (3.61 ± 0.32) × 10−5 | 319 ± 7 | 54.2 ± 2.6 | 23.0 ± 2.0 |
| 0.3 M Butanol | (4.15 ± 0.97) × 10−5 | 279 ± 10 | 59.2 ± 3.5 | 18.0 ± 4.3 |
| 0.3 mM Ascorbic acid * | (8.04 ± 0.55) × 10−5 | 144 ± 8 | 82.4 ± 4.2 | 25.6 ± 2.9 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ahmed, N.; Martienssen, M.; Bryant, I.M.; Vione, D.; Bruzzoniti, M.C.; Riedel, R. Investigation on UV Degradation and Mechanism of 6:2 Fluorotelomer Sulfonamide Alkyl Betaine, Based on Model Compound Perfluorooctanoic Acid. ChemEngineering 2024, 8, 32. https://doi.org/10.3390/chemengineering8020032
Ahmed N, Martienssen M, Bryant IM, Vione D, Bruzzoniti MC, Riedel R. Investigation on UV Degradation and Mechanism of 6:2 Fluorotelomer Sulfonamide Alkyl Betaine, Based on Model Compound Perfluorooctanoic Acid. ChemEngineering. 2024; 8(2):32. https://doi.org/10.3390/chemengineering8020032
Chicago/Turabian StyleAhmed, Naveed, Marion Martienssen, Isaac Mbir Bryant, Davide Vione, Maria Concetta Bruzzoniti, and Ramona Riedel. 2024. "Investigation on UV Degradation and Mechanism of 6:2 Fluorotelomer Sulfonamide Alkyl Betaine, Based on Model Compound Perfluorooctanoic Acid" ChemEngineering 8, no. 2: 32. https://doi.org/10.3390/chemengineering8020032