
Rate Equations of Structure-Sensitive Catalytic Reactions with Arbitrary Kinetics


Laboratory of Industrial Chemistry and Reaction Engineering Åbo Akademi University, 20500 Turku/Åbo, Finland
ChemEngineering 2023, 7(1), 12; https://doi.org/10.3390/chemengineering7010012
Submission received: 19 December 2022
/
Revised: 2 February 2023
/
Accepted: 2 February 2023
/
Published: 8 February 2023
{kind=link}
Abstract
:A general mathematical framework for the quantitative description of the cluster size dependence in heterogeneous catalytic reactions has been developed based on an analysis of the Gibbs energy of elementary reactions. The methodology was illustrated for a generic linear sequence of elementary reactions with three steps, a multi-step mechanism of ethanol oxidation comprising linear, nonlinear and quasi-equilibria steps and a network of parallel reactions in transformations of furfural.
1. Introduction
A substantial effort has been put forward in recent years in the understanding structure-sensitive reactions, for which the turnover frequency (or the rate per exposed site) depends on the size of the metal cluster [1,2,3,4,5,6] increasing, decreasing or passing through a maximum. Such changes of turnover frequency (TOF) with the cluster size can originate from changes in the relative ratio between edges, corners and terrace atoms, which exhibit different reactivity or. because of other phenomena, influence reactivity, e.g., alterations of the electronic state or resistance to deactivation.
The treatment of structure sensitivity in [7] considered differences in the adsorption energy between edges and terraces, leading subsequently to different activities of edges and terraces in terms of reactivity. A linear free energy relationship was applied linking kinetics with thermodynamics. The initial treatment [7] and further expansions were limited to a two-step sequence and some selected mechanisms, such as the Eley–Rideal and the Langmuir–Hinshelwood [8,9,10]. For more complicated reaction mechanisms, such as a Christiansen sequence of all linear steps, apart from adsorption and desorption, the equilibrium constants as well as the rate constants in forward and reverse directions of other reaction steps were considered to be independent on the cluster size.
At the same time, the reaction mechanisms can contain not only linear but also nonlinear steps, thus making a derivation of the rate equations where the cluster size dependence is directly incorporated into the rate expressions, which is far from straightforward.
The intention of the current study is to provide a general framework for the derivation of kinetic expressions for multistep reaction mechanisms with linear and nonlinear steps and different adsorbed species on the surface.
The approach will be illustrated for a Christiansen sequence and a reaction mechanism comprising reversible, irreversible and quasi-equilibria steps. The same methodology can be applied for multi-route reactions as exemplified by a network of parallel reactions.
It should be noted that the kinetic expressions derived below rely on the direct collision model assuming that the surface diffusion of adsorbed species is fast. Moreover, in the treatment of this study, the relationship between the particle size and the structure of catalytic sites is considered to be constant, being independent of cluster size. In a more general case, the electronic states of the catalytic site may change when changing the cluster size, thus influencing the reaction kinetics and even the reaction mechanism.
2. Theoretical Framework
Let us consider first an elementary reaction of the following type
A2 + * + B* = A* + AB*
Such a reaction can correspond to the dissociative adsorption of a molecule containing two atoms (e.g., O2) in the presence of some species already present on the surface (e.g., hydroxyls).
The Gibbs energy of this step can be written
as the Gibbs energy for the formation of the molecule A2 is zero by definition.
Another example can be the recombination of two adsorbed atoms of a diatomic molecule passing through the molecular adsorbed state
2A*→A2*+*
The Gibbs energy for this reaction is obviously
When only terraces and edges [7] are considered as sites with different reactivity, the Gibbs energy of reactions described by Equations (1) or (3) are expressed in the following way
where and correspond, respectively, to the reaction on terraces and edges, while denote fractions of these surface sites with their sum equal to unity.
A more detailed analysis is possible; for example, for cubooctahedral shapes of nanoparticles distinguishing reactions on different types of faces
From a relationship between the equilibrium constants and the Gibbs energy of a reaction, it follows for Equation (5)
Or more specifically, for the elementary reaction in Equation (1)
For a slightly more general case, when the Gibbs energy of formation for one of the reactants is not equal to zero, for example, for hydroxylation of an alkane on a metal surface
the Gibbs energy can be calculated from the Gibbs energy of formation of the reactants and products.
RH + O*=ROH+ *
In should be noted that for a more convenient way of kinetic analysis, the calculations of the Gibbs energy for the catalyst formation (Equation (10) and similar expressions) should be avoided. In the subsequent analysis, the Gibbs energy of adsorption per se will be used without explicitly considering the Gibbs energy for the catalyst
Such an approach gives the following instead of Equation (10)
In Equations (11) and (12), the Gibbs energy of adsorption corresponds to the difference between the Gibbs energy of formation of the adsorbed species on the catalytic sites and the Gibbs energy of formation for the catalyst, per se.
The Gibbs energy for the reaction expressed by Equation (3) is thus
In a similar fashion, instead of Equation (2)
The rate constant of a particular reaction can be expressed, making use of the linear free energy (or Brønsted–Evans–Polanyi) relationship between the reaction constants k and the equilibrium constants K in a series of analogous elementary reactions [11,12]
where g and α (Polanyi parameter, 0 < α < 1) are constants.
k = g Kα
More specifically, for the elementary reaction in Equation (1)
where k’ is the cluster size independent rate constant and corresponds to differences in Gibbs energy of adsorption on A on terraces and edges, etc.
Similarly, for the reaction given by Equation (9)
With corresponding to differences in Gibbs energy of oxygen atom adsorption on terraces and edges, etc.
In [7], the fraction of edges was related to the cluster size when d is in nm, thus allowing the introduction of the cluster size dependence directly into the rate expressions of different types.
3. Christiansen Sequence
As an example of the utilization of the above-described methodology to the derivation of rate equations, two examples will be considered. The first one corresponds to the Christiansen sequence, containing only linear steps [13,14]. The latter implies that, on both sides of an equation of the elementary surface reactions, only one adsorbed species (or vacant sites) is present. The second example treated in the section below addresses a mechanism of ethanol oxidative dehydrogenation with several nonlinear steps. For the Christiansen sequence in Equation (18), the intermediates I1 and I2 are generated in subsequent steps 1 and 2, while in the third step the vacant sites * are recovered
1.* + A1 ↔ *I1 + B1
2.*I1 ↔ *I2
3. *I2 + A2 ↔ * + B2
A1 + A2 ↔ B1 + B2
2.*I1 ↔ *I2
3. *I2 + A2 ↔ * + B2
A1 + A2 ↔ B1 + B2
For the third step, which is essentially the same as the reaction in Equation (9), the rate expression for the forward and reverse reactions are
where are the coverage of I2 and the fraction of vacant sites; and are, respectively, concentrations of A2 and B2; are frequencies of step 3 in the forward and reverse directions; and is the Polanyi parameter of step 3.
The first step of Equation (18) in the forward and reverse directions takes the form
Finally, for the second step in Equation (18), from a general expression of the Equation (16) type
The overall expression for the three-step Christiansen sequence with linear steps containing the frequencies of steps is derived assuming the steady-state approximation for all intermediates. The detailed derivation is rather tedious, being, however, explained in detail in the literature [10,13]. For the three-step sequence, an expression for TOF takes the following form
Introducing the frequencies of steps from Equations (19) to (24) into Equation (25) results in
where
Some simplifications could be made considering that the value of the Polanyi parameter is equal to 0.5 [10], implying that , and introducing the cluster size dependence :
with
4. Kinetics of Ethanol Oxidation on Gold Catalyst
A kinetic model of ethanol oxidation (EtOH) to acetaldehyde (AcH) over gold catalyst has been proposed recently in the literature [15] based on the following sequence of steps:
1. O2 + *ΞO2* 1
2. EtOH + *ΞEtOH* 2
3. EtOH* + O2*↔EtO* + OOH* 1
4. OOH* + *→O* + OH* 1
5. O* + EtO*→AcH+* + OH* 2
6. 2OH*↔O* + H2O+* 2
7. EtOH* + O*→EtO* + OH* 1
2EtOH + O2→2AcH + 2H2O
2. EtOH + *ΞEtOH* 2
3. EtOH* + O2*↔EtO* + OOH* 1
4. OOH* + *→O* + OH* 1
5. O* + EtO*→AcH+* + OH* 2
6. 2OH*↔O* + H2O+* 2
7. EtOH* + O*→EtO* + OH* 1
2EtOH + O2→2AcH + 2H2O
In Equation (28), the stoichiometric (Horiuti) numbers of the steps are given. The overall equation corresponds to the sum of all steps multiplied by these numbers.
The model was discussed in [15], invoking DFT calculations and the experimental data. The derivation of the kinetic equation for the mechanism in Equation (28) was presented in detail in the original contribution; therefore, only the cluster size dependence will be considered below. Equation (28) exhibits a combination of reversible (steps 3 and 6), irreversible (steps 4, 5 and 7) and quasi-equilibria steps (steps 1 and 2), making this example very illustrative. The fraction of vacant sites is expressed by
While the overall rate for acetaldehyde formation is
An approach to the derivation of the kinetic equations for such a complex case could be the first to identify the expressions for the Gibbs energy of quasi-equilibria steps, giving in a particular case Equation (28) for the first two steps
Subsequently, the equilibrium constants can be described through and , which correspond to the adsorption constants for the first and second steps on terraces and parameters , etc., reflecting the difference between the Gibbs energy of dioxygen adsorption on edges and terraces, etc.
For the third step in Equation (28), not containing any species present in the reaction mixture, the Gibbs energy of the surface reaction is
Analogously, for the fourth and seventh steps
Steps 5 and 6 contain reactants, giving
The rate constants of step 3 in the forward direction is expressed directly from Equation (15)
Or
Analogously, for other steps of the similar type
where
For steps 5 and 6, in the forward direction
With
The only reversible steps in this reaction mechanism are steps (3) and (6), whose rate constants are apparently
resulting in
With
Some simplifications are possible if the values of Polanyi parameters are considered to be the same for all steps. Instead of Equation (30), an expression with the cluster size dependence is obtained
where the denominator is also dependent on the cluster size
Additionally, the terms in Equation (54) are
5. Analysis of Selectivity in Parallel Reactions
Apparently, the kinetic expression developed in the previous section is cumbersome, making the analysis of the rate dependence on the cluster size rather challenging. However, in many cases when kinetics is less complicated, the general methodology described here can be utilized quite easily. As an example, an analysis of a reaction network with two parallel routes with a common adsorption step and mechanistically different rate-determining steps (i.e., mono- and bimolecular) is presented below
1. * + H2Ξ*H2
2. * + A Ξ *A
3. *A + *H2→2* + B
4. *A→* + C + E
A + H2 -> B; A -> C + E
2. * + A Ξ *A
3. *A + *H2→2* + B
4. *A→* + C + E
A + H2 -> B; A -> C + E
This mechanism, which assumes noncompetitive adsorption of hydrogen and the reactant A, was applied in [16] for analysis of the activation energy dependence on the Ru cluster size in furfural (A) hydrogenation to furfuryl alcohol (B) and decarbonylation to furan (C) and CO2 (E) [17].
Selectivity to furfuryl alcohol is thus
The equilibrium constant of the first step is similar to the one presented above in Equation (33) for oxygen. In the case of hydrogen adsorption, it takes the form
where corresponds to the difference between the Gibbs energy of dihydrogen adsorption on edges and terraces. For the second term, the corresponding constant is apparently
From the Gibbs energy for steps 3 and 4, it follows
The corresponding rate constants are therefore (with , where the cluster size is in nm)
resulting subsequently in the expression for the cluster size dependent selectivity
As demonstrated in [16], if the adsorption terms in the denominators of Equations (61) and (62) are neglected, the dependence of selectivity to the reactants vs. the metal cluster size cannot be correctly accounted for. However, some simplifications of Equation (68) can be performed to make it more tractable. The Polanyi parameters of steps 3 and 4 can be set equal to each and, moreover, equal to 0.5, as often reported in the literature [13]. This leads to the following expression of selectivity
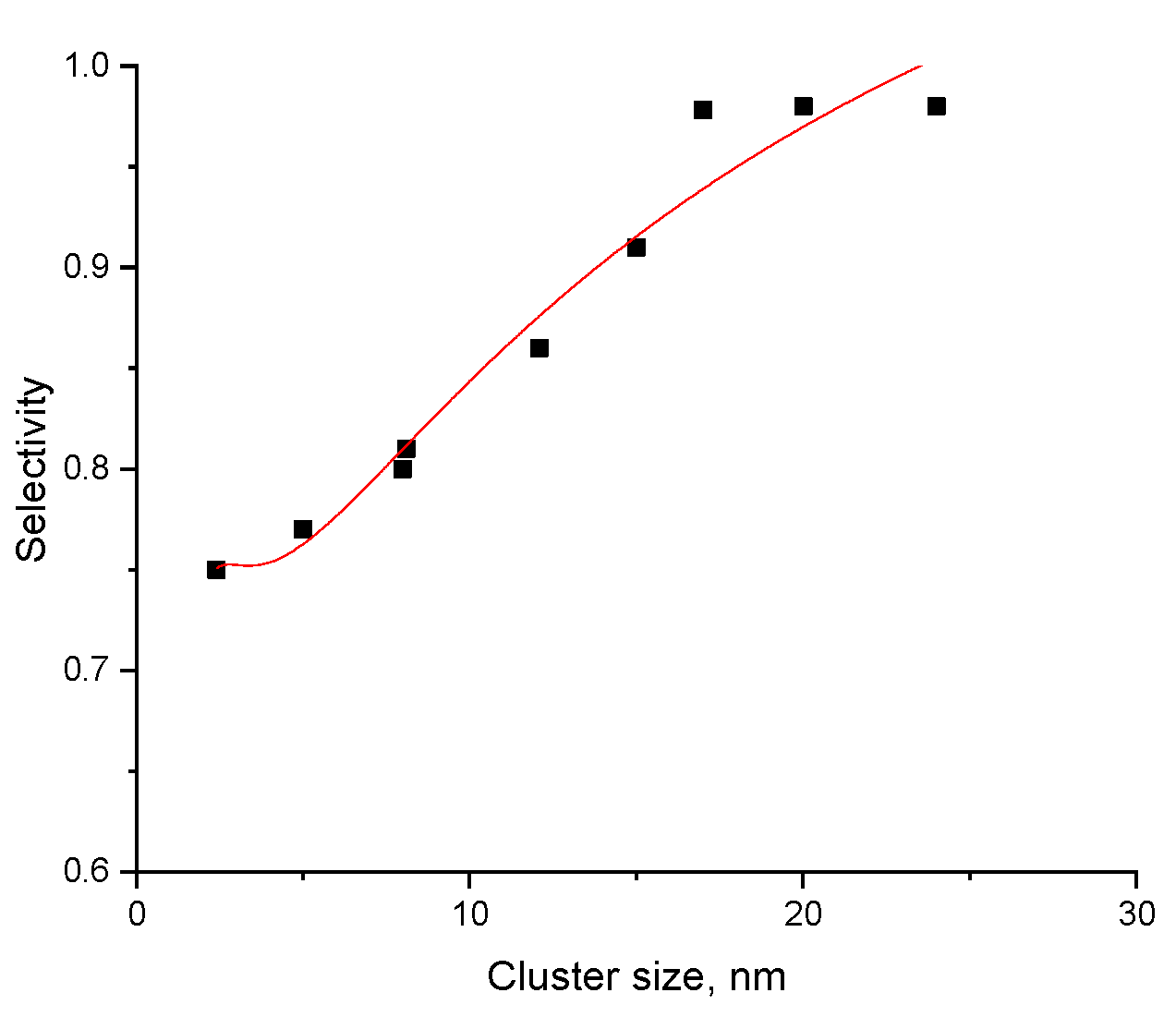
which can describe selectivity in a correct way (Figure 1).
Equation (69), although capable of an adequate description of selectivity, is over parametrized because of too many parameters, which correlate with each other. Analysis of the physico–chemical validity of the parameters based on available data cannot be properly performed. Such analysis is in any case outside of the scope of the current work aimed at presenting a general approach for deriving the rate equations of structure-sensitive reactions with arbitrary kinetics.
6. Conclusions
A general methodology for the derivation of kinetic equations in the case of structure-sensitive heterogeneous catalytic reactions was developed following the linear free energy approach for elementary steps. First, the expressions for the Gibbs energy of the elementary steps constituting the mechanism are identified considering the Gibbs energy of adsorption rather than the Gibbs energy of formation for the catalyst and the adsorbed species. This is followed by defining the equilibrium constants of the steps through the respective Gibbs energy. The Gibbs energy of adsorption for surface species is calculated through the contribution of the Gibbs energy on terraces and edges, with the fraction of edges in turn defined as a reciprocal value of the cluster size.
Finally, the rate constants of various steps are expressed as a function of the cluster size with the aid of the linear free energy relationship linking them with the equilibrium constants of the corresponding steps. The resulting rate constants are directly incorporated into the rate equations.
The methodology presented here was illustrated for the three-step generic sequence of all linear steps as well as a multistep mechanism of ethanol oxidative dehydrogenation to acetaldehyde, comprising several linear, nonlinear and quasi-equilibria steps. The resulting equations can be rather complicated, reflecting, on the other hand, the complexity of the reaction mechanisms. For much simpler cases, like the analysis of selectivity in a parallel reaction of different reaction order, the proposed methodology can be efficiently applied, demonstrating very good correspondence between the experimental data on furfural transformations used as a case study and the calculations.
Funding
This research received no external funding.
Data Availability Statement
Not applicable.
Conflicts of Interest
The author declares no conflict of interest.
References
- van Santen, R.A. Complimentary structure sensitivity and insensitive catalytic relationships. Acc. Chem. Res. 2009, 42, 57–66. [Google Scholar] [CrossRef] [PubMed]
- Vogt, C.; Kranenborg, J.; Monai, M.; Weckhuysen, B.M. Structure sensitivity in steam and dry methane reforming over nickel: Activity and carbon formation. ACS Catal. 2020, 10, 1428–1438. [Google Scholar] [CrossRef]
- Delen, G.; Monai, M.; Stanciakova, K.; Baumgartner, B.; Meirer, F.; Weckhuysen, B.M. Structure sensitivity in gas sorption and conversion on metal-organic frameworks. Nature Comm. 2023, 14, 129. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Yang, B. Structure sensitivity of ethanol steam reforming over the Rh catalyst: Reaction kinetics and deactivation mechanisms. Appl. Surf. Sci. 2023, 614, 156116. [Google Scholar] [CrossRef]
- Zong, X.; Vlachos, D.G. Exploring structure sensitivity relations for small species adsorption using machine learning. J. Chem. Inform. Model. 2022, 62, 4361–4368. [Google Scholar] [CrossRef] [PubMed]
- Murzin, D.Y. Nanokinetics for nanocatalysis. Catal. Sci. Technol. 2011, 1, 380–384. [Google Scholar]
- Murzin, D.Y. Kinetic analysis of cluster size dependent activity and selectivity. J. Catal. 2010, 276, 85–91. [Google Scholar] [CrossRef]
- Laidler, K.J. Chemical Kinetics, 3rd ed.; Harper-Collins: New York, NY, USA, 1987. [Google Scholar]
- Masel, R.J. Chemical Kinetics and Catalysis; Wiley: New York, NY, USA, 2011. [Google Scholar]
- Murzin, D.Y.; Salmi, T. Catalytic Kinetics, Science and Engineering; Elsevier: Amsterdam, The Netherlands, 2016. [Google Scholar]
- Brønsted, J.N. Acid and basic catalysis. Chem. Rev. 1928, 5, 231–338. [Google Scholar] [CrossRef]
- Arnaut, L.; Formosinho, S.; Burrows, H. Chemical Kinetics, From Molecular Structure to Chemical Reactivity; Elsevier: Amsterdam, The Netherlands, 2006. [Google Scholar]
- Temkin, M.I. The kinetics of some industrial heterogeneous catalytic reactions. Adv. Catal. 1979, 28, 173–291. [Google Scholar]
- Christiansen, J.A. The elucidation of reaction mechanisms by the method of intermediates in quasi-stationary concentrations. Adv. Catal. 1953, 5, 311–353. [Google Scholar]
- Behravesh, E.; Melander, M.M.; Wärnå, J.; Salmi, T.; Honkala, K.; Murzin, D.Y. Oxidative dehydrogenation of ethanol on gold: Combination of kinetic experiments and computation approach to unravel the reaction mechanism. J. Catal. 2021, 394, 193–205. [Google Scholar] [CrossRef]
- Murzin, D.Y. Influence of structure sensitivity on apparent activation energy of parallel heterogeneous catalytic reactions. Catal. Lett. 2020, 150, 1561–1570. [Google Scholar] [CrossRef]
- Dundell, L.J.; Zou, G.; Shangguan, W.; Lee, A.F.; Wilson, K. Structure-reactivity relations in ruthenium catalysed furfural hydrogenation. ChemCatChem 2019, 11, 3927–3932. [Google Scholar] [CrossRef]
Figure 1.
Dependence of selectivity to furfuryl alcohol in transformations of furfural over Ru on silica catalyst at 100 °C. Experimental data digitalized from [17].
Figure 1.
Dependence of selectivity to furfuryl alcohol in transformations of furfural over Ru on silica catalyst at 100 °C. Experimental data digitalized from [17].

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Murzin, D.Y. Rate Equations of Structure-Sensitive Catalytic Reactions with Arbitrary Kinetics. ChemEngineering 2023, 7, 12. https://doi.org/10.3390/chemengineering7010012
AMA Style
Murzin DY. Rate Equations of Structure-Sensitive Catalytic Reactions with Arbitrary Kinetics. ChemEngineering. 2023; 7(1):12. https://doi.org/10.3390/chemengineering7010012
Chicago/Turabian StyleMurzin, Dmitry Yu. 2023. "Rate Equations of Structure-Sensitive Catalytic Reactions with Arbitrary Kinetics" ChemEngineering 7, no. 1: 12. https://doi.org/10.3390/chemengineering7010012