

Expression Profiles of Hypoxia-Related Genes of Cancers Originating from Anatomically Similar Locations Using TCGA Database Analysis

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Selection of Hypoxia-Related Genes
2.2. Group-Specific Genetic Risk Gene Identification
2.3. Survival Analysis and Internal Validation
2.4. External Validation
2.5. Functional Enrichment Analysis
2.6. Gene Ontology Analysis
3. Results
3.1. Selection of Hypoxia-Related Genes
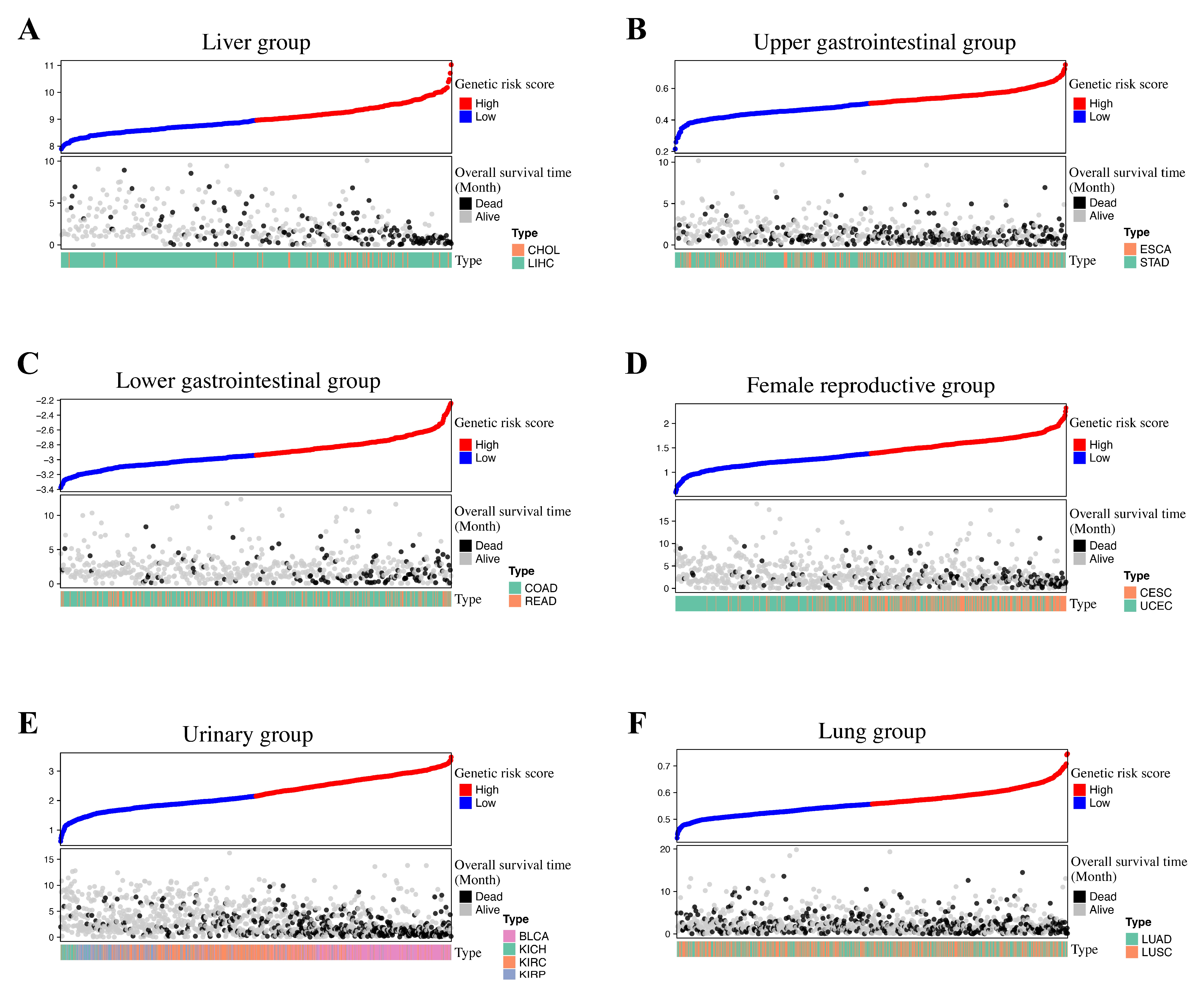
3.2. Group-Specific Genetic Risk Score Identification
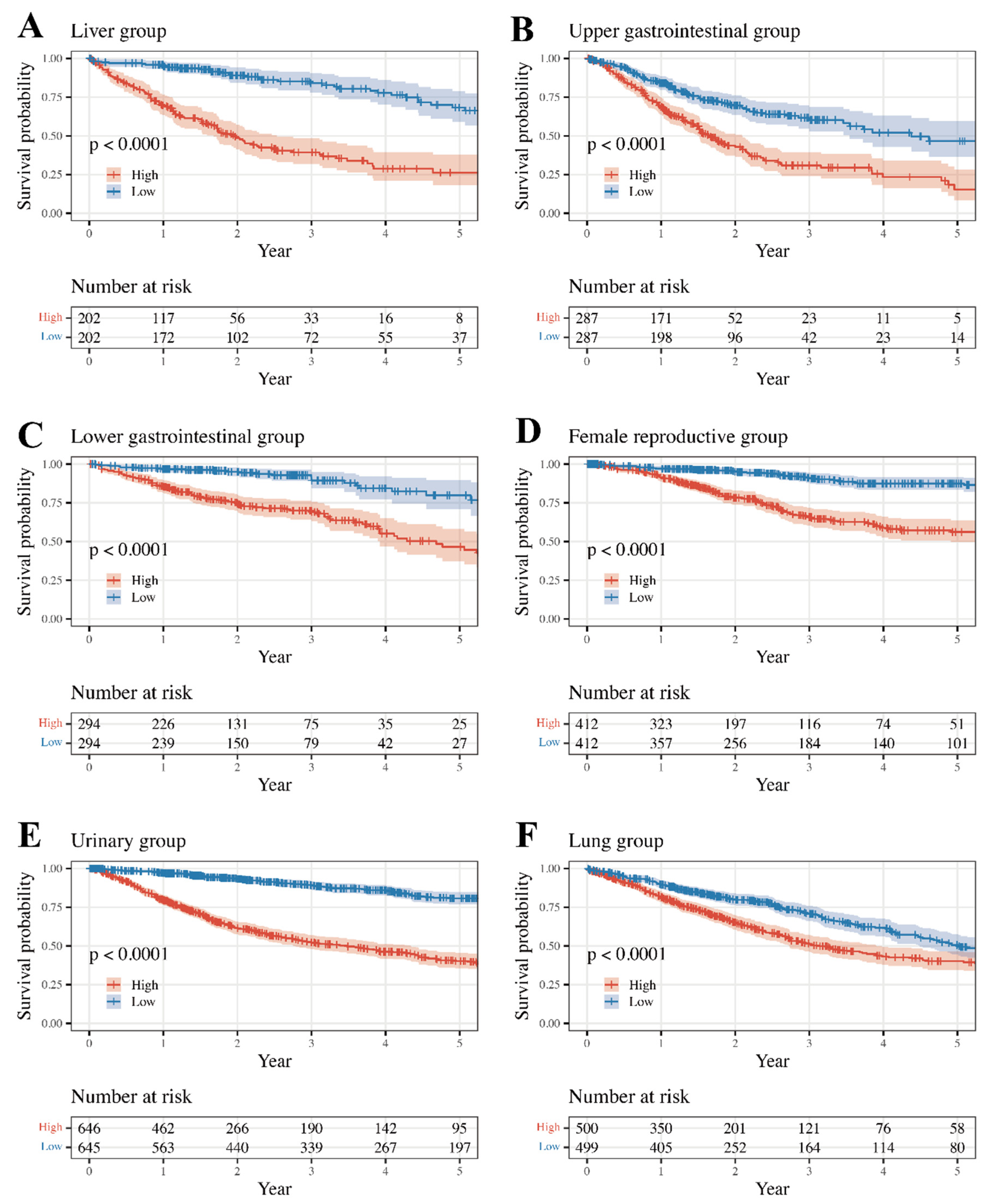
3.3. Survival Analysis and Internal Validation
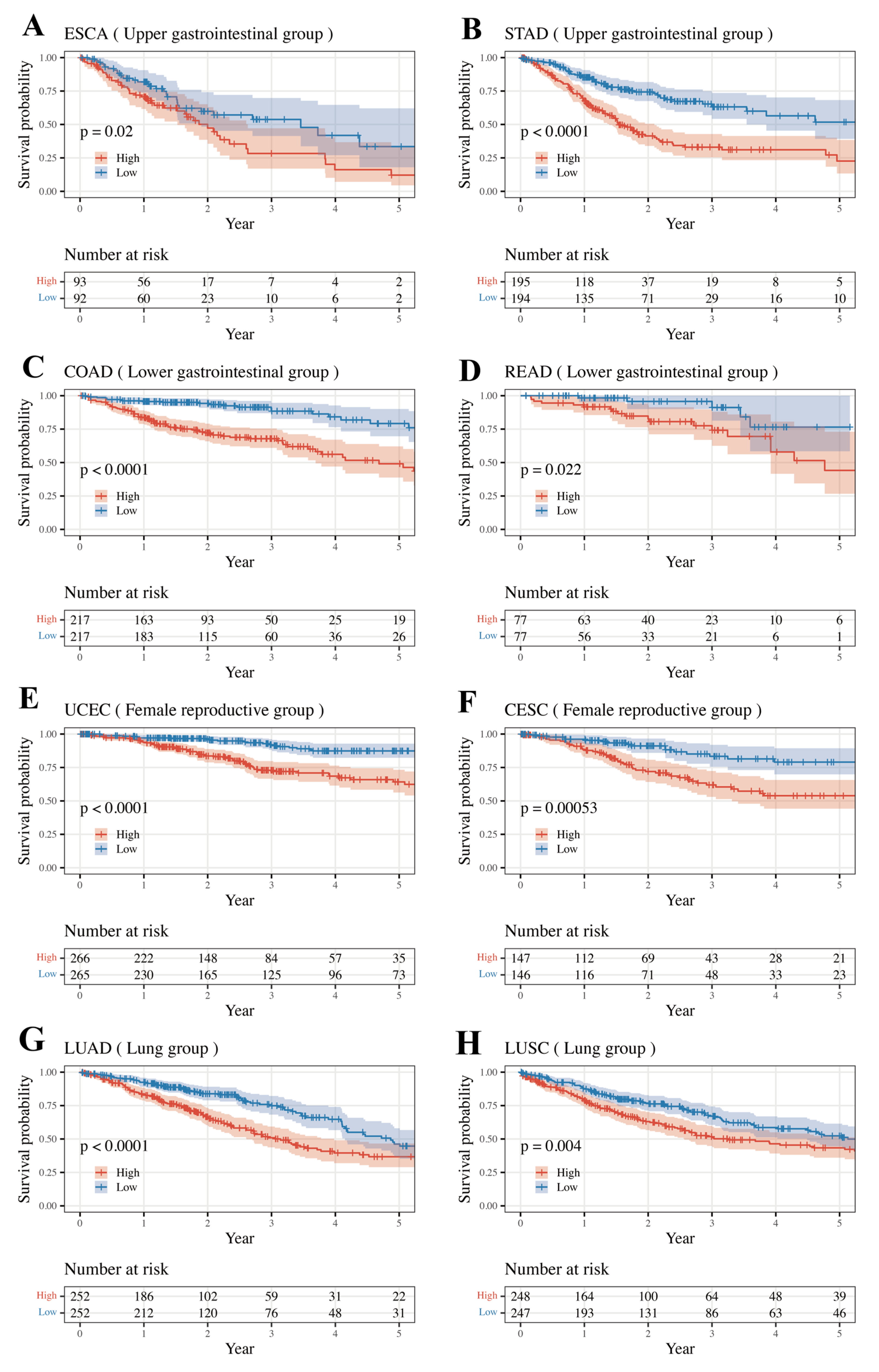
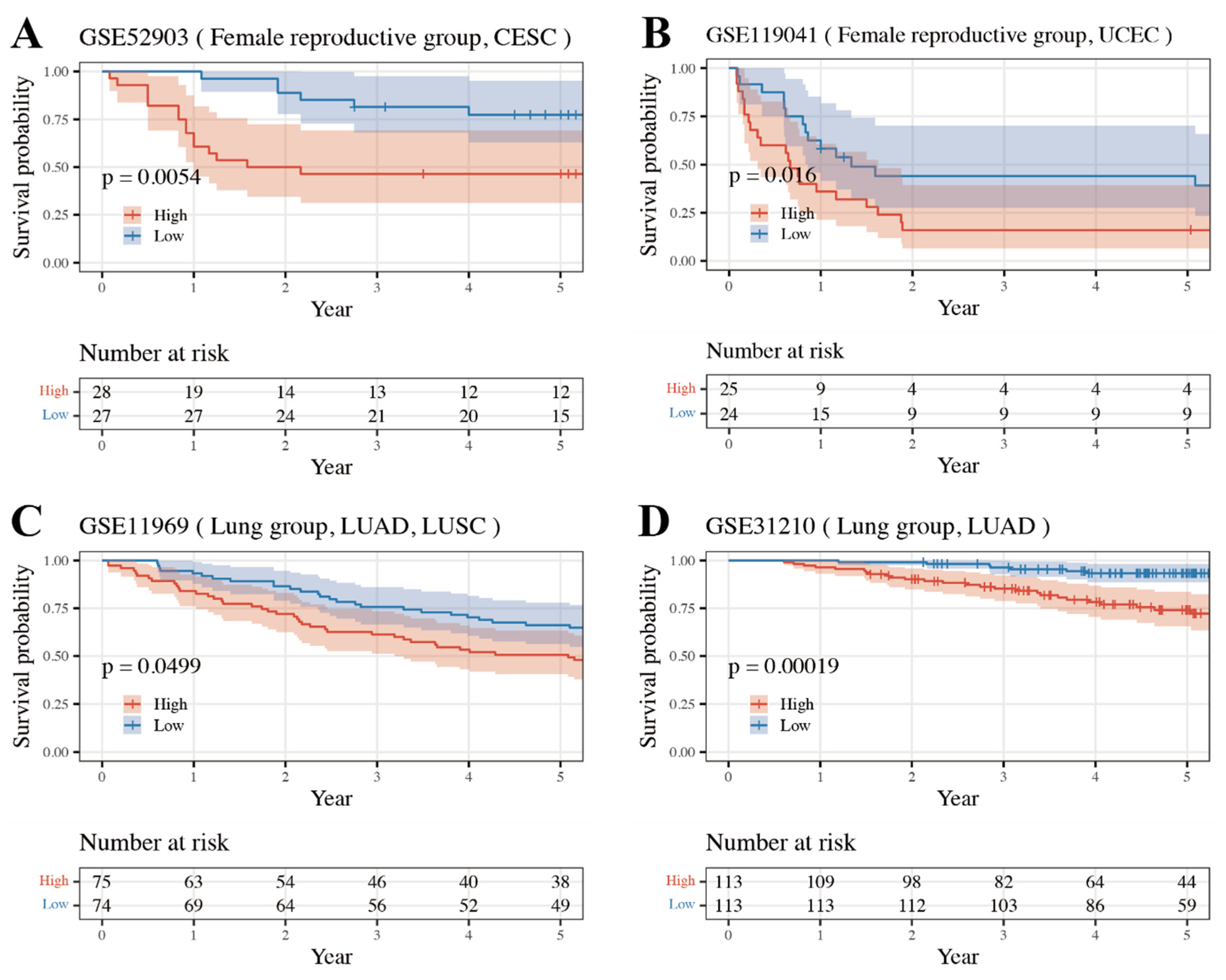
3.4. External Validation Result of Internally Validated Groups
3.5. Functional Enrichment Analysis
3.6. Gene Ontology Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Barron, C.C.; Bilan, P.J.; Tsakiridis, T.; Tsiani, E. Facilitative glucose transporters: Implications for cancer detection, prognosis and treatment. Metab. Clin. Exp. 2016, 65, 124–139. [Google Scholar] [CrossRef] [PubMed]
- D’Ignazio, L.; Batie, M.; Rocha, S. Hypoxia and Inflammation in Cancer, Focus on HIF and NF-kappaB. Biomedicines 2017, 5, 21. [Google Scholar] [CrossRef]
- Chai, Y.J.; Yi, J.W.; Oh, S.W.; Kim, Y.A.; Yi, K.H.; Kim, J.H.; Lee, K.E. Upregulation of SLC2 (GLUT) family genes is related to poor survival outcomes in papillary thyroid carcinoma: Analysis of data from the Cancer Genome Atlas. Surgery 2017, 161, 188–194. [Google Scholar] [CrossRef]
- Gan, L.; Meng, J.; Xu, M.; Liu, M.; Qi, Y.; Tan, C.; Wang, Y.; Zhang, P.; Weng, W.; Sheng, W.; et al. Extracellular matrix protein 1 promotes cell metastasis and glucose metabolism by inducing integrin β4/FAK/SOX2/HIF-1α signaling pathway in gastric cancer. Oncogene 2018, 37, 744–755. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Wang, A.; Tang, X.; Chen, Y.; Tang, E.; Jiang, H. Comparative mutational analysis of distal colon cancer with rectal cancer. Oncol. Lett. 2020, 19, 1781–1788. [Google Scholar] [CrossRef] [PubMed]
- Yerukala Sathipati, S.; Tsai, M.J.; Carter, T.; Allaire, P.; Shukla, S.K.; Beheshti, A.; Ho, S.Y. Survival estimation in patients with stomach and esophageal carcinoma using miRNA expression profiles. Comput. Struct. Biotechnol. J. 2022, 20, 4490–4500. [Google Scholar] [CrossRef] [PubMed]
- Salem, M.E.; Puccini, A.; Xiu, J.; Raghavan, D.; Lenz, H.-J.; Korn, W.M.; Shields, A.F.; Philip, P.A.; Marshall, J.L.; Goldberg, R.M. Comparative Molecular Analyses of Esophageal Squamous Cell Carcinoma, Esophageal Adenocarcinoma, and Gastric Adenocarcinoma. Oncologist 2018, 23, 1319–1327. [Google Scholar] [CrossRef]
- Zhang, Z.; Furge, K.A.; Yang, X.J.; Teh, B.T.; Hansel, D.E. Comparative gene expression profiling analysis of urothelial carcinoma of the renal pelvis and bladder. BMC Med. Genom. 2010, 3, 58. [Google Scholar] [CrossRef]
- Weinstein, J.N.; Collisson, E.A.; Mills, G.B.; Shaw, K.R.M.; Ozenberger, B.A.; Ellrott, K.; Sander, C.; Stuart, J.M.; Chang, K.; Creighton, C.J.; et al. The cancer genome atlas pan-cancer analysis project. Nat. Genet. 2013, 45, 1113–1120. [Google Scholar] [CrossRef]
- Kanehisa, M. Toward understanding the origin and evolution of cellular organisms. Protein Sci. 2019, 28, 1947–1951. [Google Scholar] [CrossRef]
- Friedman, J.; Hastie, T.; Tibshirani, R. Regularization paths for generalized linear models via coordinate descent. J. Stat. Softw. 2010, 33, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.; Hu, E.; Xu, S.; Chen, M.; Guo, P.; Dai, Z.; Feng, T.; Zhou, L.; Tang, W.; Zhan, L.; et al. ClusterProfiler 4.0: A universal enrichment tool for interpreting omics data. Innovation 2021, 2, 100141. [Google Scholar] [CrossRef] [PubMed]
- López-Cortés, A.; Guevara-Ramírez, P.; Guerrero, S.; Ortiz-Prado, E.; García-Cárdenas, J.M.; Zambrano, A.K.; Armendáriz-Castillo, I.; Pérez-Villa, A.; Yumiceba, V.; Varela, N.; et al. Metastatic signaling of hypoxia-related genes across TCGA Pan-Cancer types. bioRxiv 2020, preprint. [Google Scholar] [CrossRef]
- Guo, Y.; Liu, J.; Luo, J.; You, X.; Weng, H.; Wang, M.M.; Ouyang, T.; Li, X.; Liao, X.; Wang, M.M.; et al. Molecular Profiling Reveals Common and Specific Development Processes in Different Types of Gynecologic Cancers. Front. Oncol. 2020, 10, 584793. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.H.; Cai, J.H.; Chen, D.L.; Liao, S.H.; Lin, Y.Z.; Chung, Y.T.; Tsai, J.J.P.; Wang, C.C.N. Lasso and bioinformatics analysis in the identification of key genes for prognostic genes of gynecologic cancer. J. Pers. Med. 2021, 11, 1177. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Ye, Y.; Wang, Z.; He, Y.; Li, Y.; Mao, H.; Zhao, C. High expression of fibronectin 1 indicates poor prognosis in gastric cancer. Oncol. Lett. 2020, 19, 93–102. [Google Scholar] [CrossRef] [PubMed]
- Jalloul, A.H.; Szerencsei, R.T.; Schnetkamp, P.P.M. Cation dependencies and turnover rates of the human K+-dependent Na+-Ca2+ exchangers NCKX1, NCKX2, NCKX3 and NCKX4. Cell Calcium 2016, 59, 1–11. [Google Scholar] [CrossRef]
- Liu, J.; Feng, M.; Li, S.; Nie, S.; Wang, H.; Wu, S.; Qiu, J.; Zhang, J.; Cheng, W. Identification of molecular markers associated with the progression and prognosis of endometrial cancer: A bioinformatic study. Cancer Cell Int. 2020, 20, 59. [Google Scholar] [CrossRef]
- Zanotti, L.; Romani, C.; Tassone, L.; Todeschini, P.; Tassi, R.A.; Bandiera, E.; Damia, G.; Ricci, F.; Ardighieri, L.; Calza, S.; et al. MAL gene overexpression as a marker of high-grade serous ovarian carcinoma stem-like cells that predicts chemoresistance and poor prognosis. BMC Cancer 2017, 17, 366. [Google Scholar] [CrossRef]
- Jha, A.; Khan, Y.; Mehdi, M.; Karim, M.R.; Mehmood, Q.; Zappa, A.; Rebholz-Schuhmann, D.; Sahay, R. Towards precision medicine: Discovering novel gynecological cancer biomarkers and pathways using linked data. J. Biomed. Semant. 2017, 8, 40. [Google Scholar] [CrossRef]
- Chen, Z.; Guo, Y.; Zhao, D.; Zou, Q.; Yu, F.; Zhang, L.; Xu, L. Comprehensive Analysis Revealed that CDKN2A is a Biomarker for Immune Infiltrates in Multiple Cancers. Front. Cell Dev. Biol. 2021, 9, 808208. [Google Scholar] [CrossRef]
- Xia, L.; Zhang, W.; Gao, L. Clinical and prognostic effects of CDKN2A, CDKN2B and CDH13 promoter methylation in ovarian cancer: A study using meta-analysis and TCGA data. Biomarkers 2019, 24, 700–711. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Shen, Z. FN1 promotes thyroid carcinoma cell proliferation and metastasis by activating the NF-kappaB pathway. Protein Pept. Lett. 2022, 30, 54–64. [Google Scholar] [CrossRef]
- Xiao, Y.; Li, Y.; Tao, H.; Humphries, B.; Li, A.; Jiang, Y.; Yang, C.; Luo, R.; Wang, Z. Integrin α5 down-regulation by miR-205 suppresses triple negative breast cancer stemness and metastasis by inhibiting the Src/Vav2/Rac1 pathway. Cancer Lett. 2018, 433, 199–209. [Google Scholar] [CrossRef] [PubMed]
- Gong, C.; Yang, Z.; Wu, F.; Han, L.; Liu, Y.; Gong, W. MIR 17 inhibits ovarian cancer cell peritoneal metastasis by targeting ITGA5 and ITGB1. Oncol. Rep. 2016, 36, 2177–2183. [Google Scholar] [CrossRef] [PubMed]
- Valle-Mendiola, A.; Gutiérrez-Hoya, A.; Lagunas-Cruz, M.D.C.; Weiss-Steider, B.; Soto-Cruz, I. Pleiotropic Effects of IL-2 on Cancer: Its Role in Cervical Cancer. Mediators Inflamm. 2016, 2016, 2849523. [Google Scholar] [CrossRef]
- Yu, S.; Li, F.; Xing, S.; Zhao, T.; Peng, W.; Xue, H.H. Hematopoietic and leukemic stem cells have distinct dependence on Tcf1 and Lef1 transcription factors. J. Biol. Chem. 2016, 291, 11148–11160. [Google Scholar] [CrossRef]
- Dueñas-González, A.; Lizano, M.; Candelaria, M.; Cetina, L.; Arce, C.; Cervera, E. Epigenetics of cervical cancer. An overview and therapeutic perspectives. Mol. Cancer 2005, 4, 38. [Google Scholar] [CrossRef]
- Jobling, P.; Pundavela, J.; Oliveira, S.M.R.; Roselli, S.; Walker, M.M.; Hondermarck, H. Nerve-cancer cell cross-talk: A novel promoter of tumor progression. Cancer Res. 2015, 75, 1777–1781. [Google Scholar] [CrossRef]
- Zhao, C.M.; Hayakawa, Y.; Kodama, Y.; Muthupalani, S.; Westphalen, C.B.; Andersen, G.T.; Flatberg, A.; Johannessen, H.; Friedman, R.A.; Renz, B.W.; et al. Denervation suppresses gastric tumorigenesis. Sci. Transl. Med. 2014, 6, 250ra115. [Google Scholar] [CrossRef]
- Albo, D.; Akay, C.L.; Marshall, C.L.; Wilks, J.A.; Verstovsek, G.; Liu, H.; Agarwal, N.; Berger, D.H.; Ayala, G.E. Neurogenesis in colorectal cancer is a marker of aggressive tumor behavior and poor outcomes. Cancer 2011, 117, 4834–4845. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.L.; Zhang, Y.; Wang, J.; Chen, N.; Fang, W.; Zhong, J.; Liu, Y.; Qin, R.; Yu, X.; Sun, Z.; et al. A 17 gene panel for non-small-cell lung cancer prognosis identified through integrative epigenomic-transcriptomic analyses of hypoxia-induced epithelial–mesenchymal transition. Mol. Oncol. 2019, 13, 1490–1502. [Google Scholar] [CrossRef] [PubMed]
- Sun, F.; Yang, X.; Jin, Y.; Chen, L.; Wang, L.; Shi, M.; Zhan, C.; Shi, Y.; Wang, Q. Bioinformatics analyses of the differences between lung adenocarcinoma and squamous cell carcinoma using the Cancer Genome Atlas expression data. Mol. Med. Rep. 2017, 16, 609–616. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Kim, H.S.; Kim, B.J.; Han, B.; Choi, D.R.; Kwon, J.H. Prognostic impact of TTF-1 expression in non-squamous non-small-cell lung cancer: A meta-analysis. J. Cancer 2018, 9, 4279–4286. [Google Scholar] [CrossRef]
- Karachaliou, N.; Rosell, R.; Viteri, S. The role of SOX2 in small cell lung cancer, lung adenocarcinoma and squamous cell carcinoma of the lung. Transl. Lung Cancer Res. 2013, 2, 172–179. [Google Scholar]
- Klahan, S.; Huang, W.C.; Chang, C.M.; Wong, H.S.; Huang, C.C.; Wu, M.S.; Lin, Y.C.; Lu, H.F.; Hou, M.F.; Chang, W.C. Gene expression profiling combined with functional analysis identify integrin beta1 (ITGB1) as a potential prognosis biomarker in triple negative breast cancer. Pharmacol. Res. 2016, 104, 31–37. [Google Scholar] [CrossRef]
- Zhang, J.; Liu, K.; Peng, P.; Li, S.; Ye, Z.; Su, Y.; Liu, S.; Qin, M.; Huang, J. Upregulation of nectin-4 is associated with ITGB1 and vasculogenic mimicry and may serve as a predictor of poor prognosis in colorectal cancer. Oncol. Lett. 2019, 18, 1163–1170. [Google Scholar] [CrossRef]
- Hou, X.; Shi, X.; Zhang, W.; Li, D.; Hu, L.; Yang, J.; Zhao, J.; Wei, S.; Wei, X.; Ruan, X.; et al. LDHA induces EMT gene transcription and regulates autophagy to promote the metastasis and tumorigenesis of papillary thyroid carcinoma. Cell Death Dis. 2021, 12, 347. [Google Scholar] [CrossRef]
- Xiao, X.; Huang, X.; Ye, F.; Chen, B.; Song, C.; Wen, J.; Zhang, Z.; Zheng, G.; Tang, H.; Xie, X. The miR-34a-LDHA axis regulates glucose metabolism and tumor growth in breast cancer. Sci. Rep. 2016, 6, 21735. [Google Scholar] [CrossRef]
- Zhang, C.; Liu, J.; Zhao, Y.; Yue, X.; Zhu, Y.; Wang, X.; Wu, H.; Blanco, F.; Li, S.; Bhanot, G.; et al. Glutaminase 2 is a novel negative regulator of small GTPase Rac1 and mediates p53 function in suppressing metastasis. Elife 2016, 5, e10727. [Google Scholar] [CrossRef]
- Lopez de la Oliva, A.R.; Campos-Sandoval, J.A.; Gomez-Garcia, M.C.; Cardona, C.; Martin-Rufian, M.; Sialana, F.J.; Castilla, L.; Bae, N.; Lobo, C.; Penalver, A.; et al. Nuclear Translocation of Glutaminase GLS2 in Human Cancer Cells Associates with Proliferation Arrest and Differentiation. Sci. Rep. 2020, 10, 2259. [Google Scholar] [CrossRef] [PubMed]
- Peng, W.J.; He, Q.; Yang, J.X.; Wang, B.X.; Lu, M.M.; Wang, S.; Wang, J. Meta-analysis of association between cytokine gene polymorphisms and lung cancer risk. Mol. Biol. Rep. 2012, 39, 5187–5194. [Google Scholar] [CrossRef] [PubMed]
- Domagala-Kulawik, J. The role of the immune system in non-small cell lung carcinoma and potential for therapeutic intervention. Transl. Lung Cancer Res. 2015, 4, 177–190. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Zhao, H. Tryptophan and Its Metabolites in Lung Cancer: Basic Functions and Clinical Significance. Front. Oncol. 2021, 11, 707277. [Google Scholar] [CrossRef]
- Fahrmann, J.F.; Vykoukal, J.V.; Ostrin, E.J. Amino Acid Oncometabolism and Immunomodulation of the Tumor Microenvironment in Lung Cancer. Front. Oncol. 2020, 10, 276. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
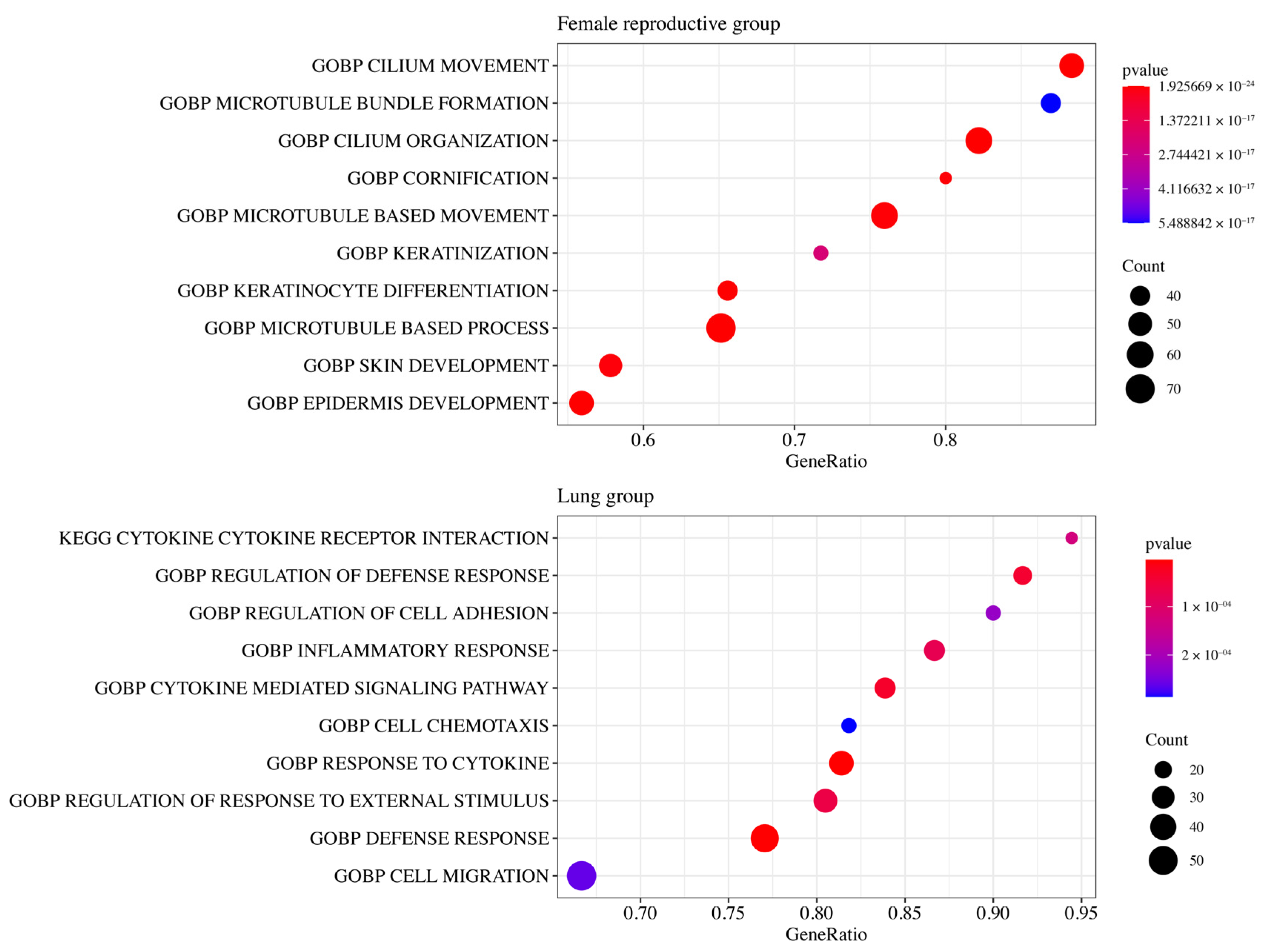{kind=link}
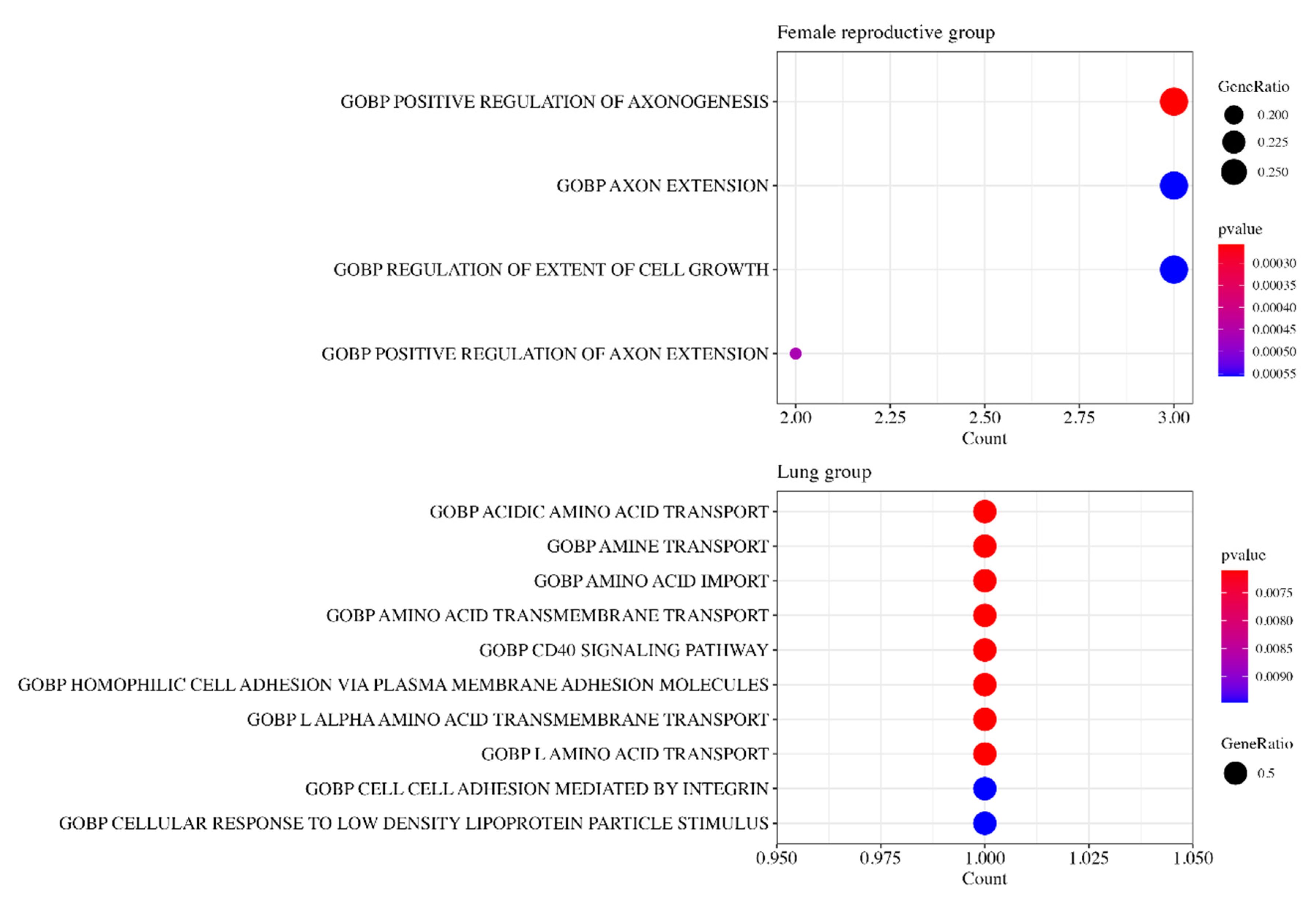{kind=link}
| Database | Pathways |
|---|---|
| KEGG | Pathways in cancer |
| Central carbon metabolism in cancer | |
| PI3K-Akt signaling pathway | |
| HIF-1 signaling pathway | |
| VEGF signaling pathway | |
| TNF signaling pathway | |
| Reactome | Hexose transport |
| Signaling by NOTCH1 | |
| MAPK targets/Nuclear events mediated by MAP kinases |
| Group | Type (Abbreviation, Number of Samples) |
|---|---|
| Liver | Liver hepatocellular carcinoma (LIHC, 368) Cholangiocarcinoma (CHOL, 36) |
| Upper Gastrointestinal | Esophageal carcinoma (ESCA, 185) Stomach adenocarcinoma (STAD, 389) |
| Lower Gastrointestinal | Colon adenocarcinoma (COAD, 434) Rectum adenocarcinoma (READ, 154) |
| Female Reproductive | Uterine corpus endometrial carcinoma (UCEC, 531) Cervical squamous cell carcinoma and endocervical adenocarcinoma (CESC, 293) |
| Urinary | Bladder urothelial carcinoma (BLCA, 406) Kidney renal clear cell carcinoma (KIRC, 532) Kidney renal papillary cell carcinoma (KIRP, 288) Kidney chromophobe (KICH, 65) |
| Lung | Lung adenocarcinoma (LUAD, 504) Lung squamous cell carcinoma (LUSC, 495) |
| Group | Group-Specific Log-Rank p-Value | Type | Type-Specific Log-Rank p-Value | Internal Validated |
|---|---|---|---|---|
| Liver | 5.12 × 10−18 | LIHC | 2.92 × 10−17 | FALSE |
| CHOL | 6.17 × 10−2 | |||
| Upper Gastrointestinal | 1.39 × 10−9 | ESCA | 1.99 × 10−2 | TRUE |
| STAD | 7.88 × 10−8 | |||
| Lower Gastrointestinal | 8.02 × 10−11 | COAD | 5.20 × 10−9 | TRUE |
| READ | 2.19 × 10−2 | |||
| Female Reproductive | 9.10 × 10−16 | UCEC | 1.51 × 10−7 | TRUE |
| CESC | 5.32 × 10−4 | |||
| Urinary | 9.86 × 10−47 | BLCA | 3.46 × 10−5 | FALSE |
| KIRC | 3.75 × 10−13 | |||
| KIRP | 7.53 × 10−5 | |||
| KICH | 8.29 × 10−2 | |||
| Lung | 1.51 × 10−6 | LUAD | 7.06 × 10−5 | TRUE |
| LUSC | 3.95 × 10−3 |
| Group | Positive Risk Coefficient | Negative Risk Coefficient |
|---|---|---|
| Liver | BIRC5, BIRC8, CUL2, EIF4E, EPO, G6PD, GNA12, HDAC1, HDAC2, HSP90AA1, IFNA13, IL8, LDHA, MAPK7, NUP155, PGF, PPP2R5B, RHEB, SLC1A5, SLC2A1, SPP1, YWHAB | CCNA1, CNTN1, FLT3, G6PC2, GHR, HES5, IFNA2, ITGB7, NTRK1, PFKL, TNF, TP53, WNT1 |
| Upper Gastrointestinal | APH1B, SERPINE1, SLC2A3, SOCS3, TF | DAB2IP, MKNK2 |
| Lower Gastrointestinal | APC2, ENO3, HEYL, TIMP1, WNT10B | CTNNA1, MAPKAPK3, TMEM48 |
| Female Reproductive | BDKRB1, CDKN2A, FN1, ITGA5, PFKM, SLC45A3, TFRC, VEGFA, WNT3, YWHAB, YWHAG | CD19, IL2RB, JMJD7-PLA2G4B, LEF1, MDM2, RBPJ |
| Urinary | BIRC5, CCNE2, COL6A3, DVL3, EIF4EBP1, FGF5, GLI2, PPP2R2C, SLC7A5, THBS3 | DAB2IP, ITGB7 |
| Lung | ITGB1, LDHA | GLS2 |
| Group | Matched TCGA Type | GEO Accession Number |
|---|---|---|
| Upper Gastrointestinal | ESCA | GSE72873 |
| STAD | GSE15459 * | |
| Lower Gastrointestinal | COAD READ | GSE41258, GSE17538, GSE72970, GSE17537, GSE17536 |
| Female Reproductive | UCEC | GSE119041 * |
| CESC | GSE52903 * | |
| Urinary | BLCA | GSE31684, GSE13507, GSE19423 |
| KIRC | GSE29609 | |
| Lung | LUAD/SC | GSE11969 *, GSE37745 |
| LUAD | GSE31210 *, GSE30219, GSE50081, GSE29014 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bae, H.L.; Jeong, K.; Yang, S.; Jun, H.; Kim, K.; Chai, Y.J. Expression Profiles of Hypoxia-Related Genes of Cancers Originating from Anatomically Similar Locations Using TCGA Database Analysis. Medicines 2024, 11, 2. https://doi.org/10.3390/medicines11010002
Bae HL, Jeong K, Yang S, Jun H, Kim K, Chai YJ. Expression Profiles of Hypoxia-Related Genes of Cancers Originating from Anatomically Similar Locations Using TCGA Database Analysis. Medicines. 2024; 11(1):2. https://doi.org/10.3390/medicines11010002
Chicago/Turabian StyleBae, Hye Lim, Kyeonghun Jeong, Suna Yang, Hyeji Jun, Kwangsoo Kim, and Young Jun Chai. 2024. "Expression Profiles of Hypoxia-Related Genes of Cancers Originating from Anatomically Similar Locations Using TCGA Database Analysis" Medicines 11, no. 1: 2. https://doi.org/10.3390/medicines11010002