
AhR Mediated Activation of Pro-Inflammatory Response of RAW 264.7 Cells Modulate the Epithelial-Mesenchymal Transition
 , , , and
, , , and
Abstract
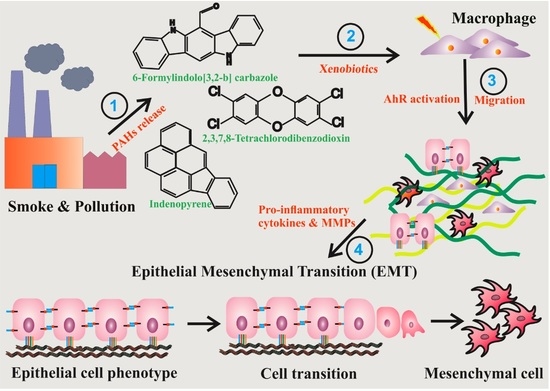
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Background
2. Materials and Methods
2.1. Chemicals, Reagents and Antibodies
2.2. Cell Line Treatment
2.3. Chemokine-Cytokine Screening
2.4. Enzyme-Linked Immunosorbent Assay
2.5. Cell—Migration Assay
2.6. Crystal Violet Staining
2.7. Zymography
2.8. Western Blotting
2.9. Statistics
3. Results
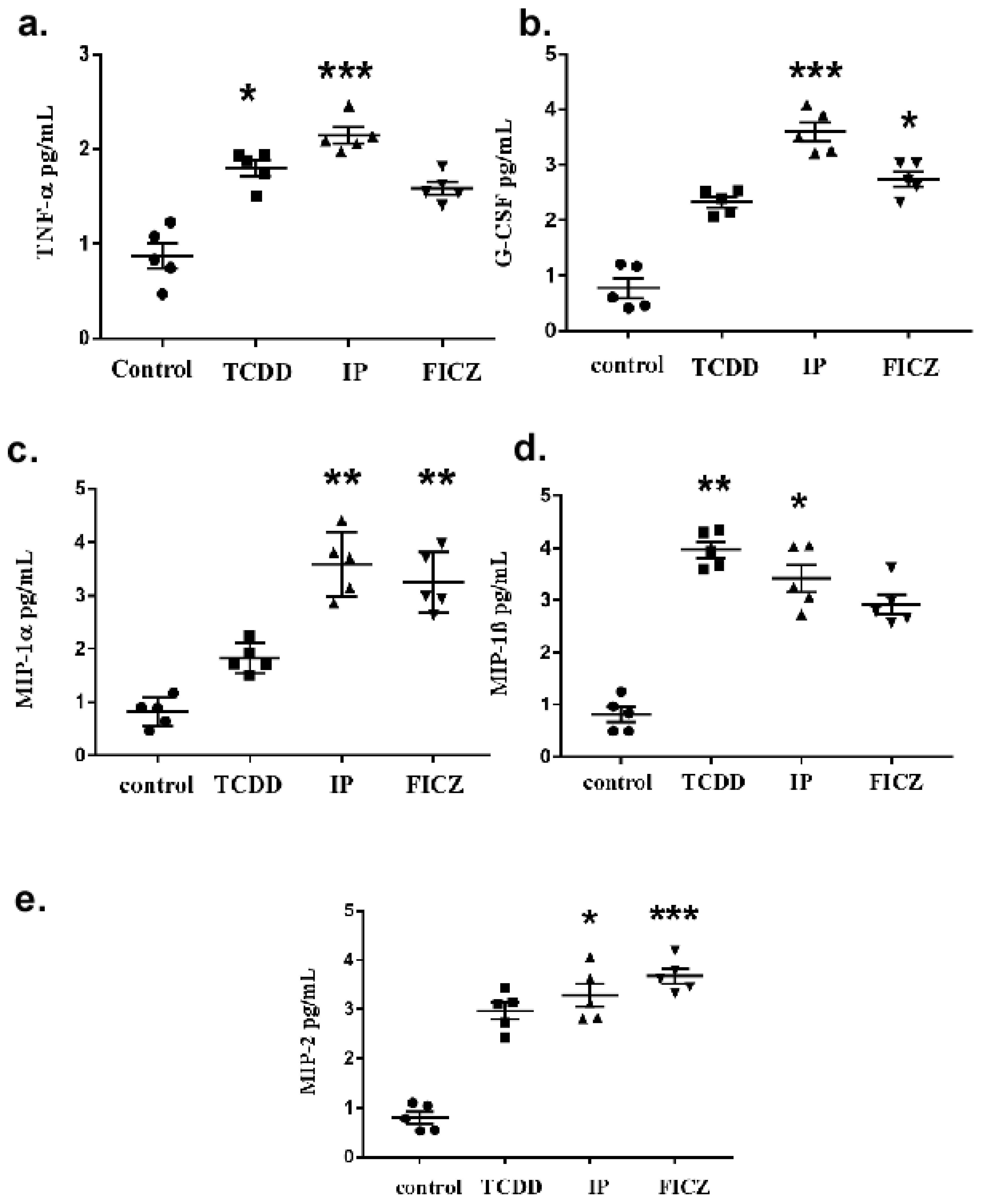
3.1. TCDD Promotes Expression of Cytokine and Chemokine in Macrophage RAW 264.7 Cells
3.2. AhR Activation by Agonists Promotes the Production of Cytokines
3.3. RAW 264.7 Cell-Conditioned Medium Induces Mouse Lung Epithelial Cell Migration
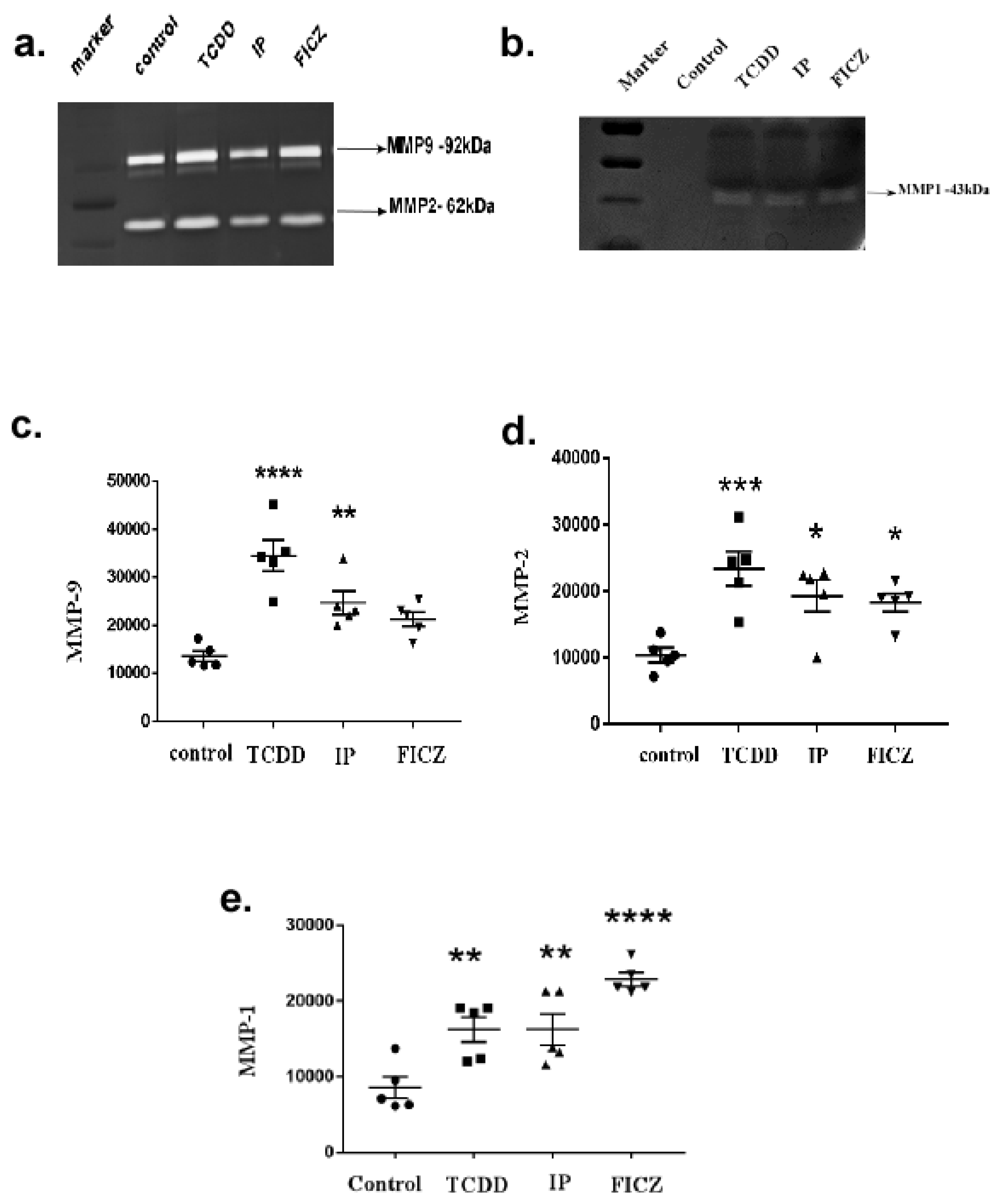
3.4. Activation of AhR Induces MMP Expression in Macrophage RAW264.7 Cells
3.5. AhR Activation Induces EMT Marker Expression in Mouse Lung Epithelial Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Cui, Z.; Feng, Y.; Li, D.; Li, T.; Gao, P.; Xu, T. Activation of aryl hydrocarbon receptor (AhR) in mesenchymal stem cells modulates macrophage polarization in asthma. J. Immunotoxicol. 2020, 17, 21–30. [Google Scholar] [CrossRef]
- Shi, Y.; Zeng, Z.; Yu, J.; Tang, B.; Tang, R.; Xiao, R. The aryl hydrocarbon receptor: An environmental effector in the pathogenesis of fibrosis. Pharmacol. Res. 2020, 160, 105180. [Google Scholar] [CrossRef]
- Reyfman, P.A.; Walter, J.M.; Joshi, N.; Anekalla, K.R.; McQuattie-Pimentel, A.C.; Chiu, S.; Fernandez, R.; Akbarpour, M.; Chen, C.-I.; Ren, Z.; et al. Single-Cell Transcriptomic Analysis of Human Lung Provides Insights into the Pathobiology of Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 2019, 199, 1517–1536. [Google Scholar] [CrossRef] [PubMed]
- Richeldi, L.; Collard, H.R.; Jones, M.G. Idiopathic pulmonary fibrosis. Lancet 2017, 389, 1941–1952. [Google Scholar] [CrossRef]
- Ding, Q.; Sun, J.; Xie, W.; Zhang, M.; Zhang, C.; Xu, X. Stemona alkaloids suppress the positive feedback loop between M2 polarization and fibroblast differentiation by inhibiting JAK2/STAT3 pathway in fibroblasts and CXCR4/PI3K/AKT1 pathway in macrophages. Int. Immunopharmacol. 2019, 72, 385–394. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Gao, J.; Qin, C.; Liang, Y.; Chen, S.; Hei, F. Inflammatory alveolar macrophage-derived microvesicles damage lung epithelial cells and induce lung injury. Immunol. Lett. 2022, 241, 23–34. [Google Scholar] [CrossRef] [PubMed]
- Hou, J.; Shi, J.; Chen, L.; Lv, Z.; Chen, X.; Cao, H.; Xiang, Z.; Han, X. M2 macrophages promote myofibroblast differentiation of LR-MSCs and are associated with pulmonary fibrogenesis. Cell Commun. Signal. 2018, 16, 89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arora, S.; Dev, K.; Agarwal, B.; Das, P.; Syed, M.A. Macrophages: Their role, activation and polarization in pulmonary diseases. Immunobiology 2018, 223, 383–396. [Google Scholar] [CrossRef]
- Ye, G.; Gao, H.; Zhang, X.; Liu, X.; Chen, J.; Liao, X.; Zhang, H.; Huang, Q. Aryl hydrocarbon receptor mediates benzo[a]pyrene-induced metabolic reprogramming in human lung epithelial BEAS-2B cells. Sci. Total Environ. 2021, 756, 144130. [Google Scholar] [CrossRef] [PubMed]
- Lamb, C.L.; Cholico, G.N.; Perkins, D.E.; Fewkes, M.T.; Oxford, J.; Lujan, T.J.; Morrill, E.E.; Mitchell, K.A. Aryl Hydrocarbon Receptor Activation by TCDD Modulates Expression of Extracellular Matrix Remodeling Genes during Experimental Liver Fibrosis. BioMed Res. Int. 2016, 2016, 5309328. [Google Scholar] [CrossRef] [PubMed]
- Murray, P.J. Macrophage Polarization. Annu. Rev. Physiol. 2017, 79, 541–566. [Google Scholar] [CrossRef] [PubMed]
- Lim, H.; Park, J.-Y.; Abekura, F.; Choi, H.; Kim, H.-D.; Magae, J.; Chang, Y.-C.; Lee, Y.-C.; Kim, C.-H. 4-O-methylascochlorin attenuates inflammatory responses induced by lipopolysaccharide in RAW 264.7 macrophages. Int. Immunopharmacol. 2021, 90, 107184. [Google Scholar] [CrossRef] [PubMed]
- Kuo, C.-Y.; Yang, T.-H.; Tsai, P.-F.; Yu, C.-H. Role of the Inflammatory Response of RAW 264.7 Cells in the Metastasis of Novel Cancer Stem-Like Cells. Medicina 2021, 57, 778. [Google Scholar] [CrossRef]
- Zhang, W.-J.; Wang, X.-H.; Gao, S.-T.; Chen, C.; Xu, X.-Y.; Sun, Q.; Zhou, Z.-H.; Wu, G.-Z.; Yu, Q.; Xu, G.; et al. Tumor-associated macrophages correlate with phenomenon of epithelial-mesenchymal transition and contribute to poor prognosis in triple-negative breast cancer patients. J. Surg. Res. 2018, 222, 93–101. [Google Scholar] [CrossRef]
- Lou, W.; Zhang, M.-D.; Chen, Q.; Bai, T.-Y.; Hu, Y.-X.; Gao, F.; Li, J.; Lv, X.-L.; Zhang, Q.; Chang, F.-H. Molecular mechanism of benzo [a] pyrene regulating lipid metabolism via aryl hydrocarbon receptor. Lipids Health Dis. 2022, 21, 13. [Google Scholar] [CrossRef]
- Cheon, H.J.; Woo, Y.-S.; Lee, J.Y.; Kim, H.S.; Kim, H.J.; Cho, S.; Won, N.H.; Sohn, W. Signaling pathway for 2,3,7,8-tetrachlorodibenzo- p-dioxin-induced TNF-α production in differentiated THP-1 human macrophages. Exp. Mol. Med. 2007, 39, 524–534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Facchin, B.M.; dos Reis, G.O.; Vieira, G.N.; Mohr, E.T.B.; da Rosa, J.S.; Kretzer, I.F.; Demarchi, I.G.; Dalmarco, E.M. Inflammatory biomarkers on an LPS-induced RAW 264.7 cell model: A systematic review and meta-analysis. Inflamm. Res. 2022, 71, 741–758. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Ding, W.; Deng, X. PM2.5, Fine Particulate Matter: A Novel Player in the Epithelial-Mesenchymal Transition? Front. Physiol. 2019, 10, 1404. [Google Scholar] [CrossRef] [PubMed]
- Larigot, L.; Benoit, L.; Koual, M.; Tomkiewicz, C.; Barouki, R.; Coumoul, X. Aryl Hydrocarbon Receptor and Its Diverse Ligands and Functions: An Exposome Receptor. Annu. Rev. Pharmacol. Toxicol. 2022, 62, 383–404. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Sha, R.; Xu, L.; Xia, Y.; Liu, Y.; Li, X.; Xie, H.Q.; Tang, N.; Zhao, B. 2,3,7,8-Tetrachlorodibenzo-p-dioxin promotes migration ability of primary cultured rat astrocytes via aryl hydrocarbon receptor. J. Environ. Sci. 2019, 76, 368–376. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.C.; Oslund, K.L.; Thai, P.; Velichko, S.; Fujisawa, T.; Duong, T.; Denision, M.S.; Wu, R. 2,3,7,8-Tetrachlorodibenzo-p-dioxin-induced MUC5AC expression: Aryl hydrocarbon receptor-independent/EGFR/ERK/p38-dependent SP1-based transcription. Am. J. Respir. Cell Mol. Biol. 2011, 45, 270–276. [Google Scholar] [CrossRef] [Green Version]
- Esteban, J.; Sánchez-Pérez, I.; Hamscher, G.; Miettinen, H.M.; Korkalainen, M.; Viluksela, M.; Pohjanvirta, R.; Håkansson, H. Role of aryl hydrocarbon receptor (AHR) in overall retinoid metabolism: Response comparisons to 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) exposure between wild-type and AHR knockout mice. Reprod. Toxicol. 2021, 101, 33–49. [Google Scholar] [CrossRef] [PubMed]
- Hammond, C.L.; Roztocil, E.; Phipps, R.P.; Feldon, S.E.; Woeller, C.F. Proton pump inhibitors attenuate myofibroblast formation associated with thyroid eye disease through the aryl hydrocarbon receptor. PLoS ONE 2019, 14, e0222779. [Google Scholar] [CrossRef] [Green Version]
- Su, H.-H.; Lin, H.-T.; Suen, J.-L.; Sheu, C.C.; Yokoyama, K.K.; Huang, S.-K.; Cheng, C.M. Aryl hydrocarbon receptor–ligand axis mediates pulmonary fibroblast migration and differentiation through increased arachidonic acid metabolism. Toxicology 2016, 370, 116–126. [Google Scholar] [CrossRef] [PubMed]
- Walia, V.; Samuels, Y. Analysis of Enzymatic Activity of Matrix Metalloproteinase (MMP) by Collagen Zymography in Melanoma. Proteases and Cancer. Methods Mol. Biol. 2018, 173, 97–106. [Google Scholar]
- Vogel, C.F.A.; Ishihara, Y.; Campbell, C.E.; Kado, S.Y.; Nguyen-Chi, A.; Sweeney, C.; Pollet, M.; Haarmann-Stemmann, T.; Tuscano, J.M. A Protective Role of Aryl Hydrocarbon Receptor Repressor in Inflammation and Tumor Growth. Cancers 2019, 11, 589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murray, I.A.; Perdew, G.H. How Ah Receptor Ligand Specificity Became Important in Understanding Its Physiological Function. Int. J. Mol. Sci. 2020, 21, 9614. [Google Scholar] [CrossRef] [PubMed]
- Wong, T.-H.; Lee, C.-L.; Su, H.-H.; Lee, C.-L.; Wu, C.-C.; Wang, C.-C.; Sheu, C.-C.; Lai, R.-S.; Leung, S.-Y.; Lin, C.-C.; et al. A prominent air pollutant, Indeno[1,2,3-cd]pyrene, enhances allergic lung inflammation via aryl hydrocarbon receptor. Sci. Rep. 2018, 8, 5198. [Google Scholar] [CrossRef] [Green Version]
- Scheraga, R.G.; Thannickal, V.J. Wnt/beta-catenin and transforming growth factor-beta signaling in pulmonary fibrosis. A case for antagonistic pleiotropy? Am. J. Respir. Crit. Care Med. 2014, 190, 129–131. [Google Scholar] [CrossRef] [PubMed]
- Ianni, A.; Hofmann, M.; Kumari, P.; Tarighi, S.; Al-Tamari, H.M.; Gorgens, A.; Giebel, B.; Nolte, H.; Kruger, M.; Salwig, I.; et al. Depletion of Numb and Numblike in Murine Lung Epithelial Cells Ameliorates Bleomycin-Induced Lung Fibrosis by Inhibiting the beta-Catenin Signaling Pathway. Front. Cell Dev. Biol. 2021, 9, 639162. [Google Scholar] [CrossRef]
- Shi, J.; Li, F.; Luo, M.; Wei, J.; Liu, X. Distinct Roles of Wnt/β-Catenin Signaling in the Pathogenesis of Chronic Obstructive Pulmonary Disease and Idiopathic Pulmonary Fibrosis. Mediat. Inflamm. 2017, 2017, 3520581. [Google Scholar] [CrossRef]
- Cao, H.; Wang, C.; Chen, X.; Hou, J.; Xiang, Z.; Shen, Y.; Han, X. Inhibition of Wnt/β-catenin signaling suppresses myofibroblast differentiation of lung resident mesenchymal stem cells and pulmonary fibrosis. Sci. Rep. 2018, 8, 13644. [Google Scholar] [CrossRef] [Green Version]
- Ota, C.; Baarsma, H.A.; Wagner, D.E.; Hilgendorff, A.; Königshoff, M. Linking bronchopulmonary dysplasia to adult chronic lung diseases: Role of WNT signaling. Mol. Cell. Pediatr. 2016, 3, 34. [Google Scholar] [CrossRef] [Green Version]
- Shiizaki, K.; Kido, K.; Mizuta, Y. Insight into the relationship between aryl-hydrocarbon receptor and β-catenin in human colon cancer cells. PLoS ONE 2019, 14, e0224613. [Google Scholar] [CrossRef]
- Singleman, C.; Holtzman, N.G. PCB and TCDD derived embryonic cardiac defects result from a novel AhR pathway. Aquat. Toxicol. 2021, 233, 105794. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Li, Y.; Zhou, R.; Wang, N.; Kang, S. Knockdown of E-cadherin expression of endometrial epithelial cells may activate Wnt/β-catenin pathway in vitro. Arch. Gynecol. Obstet. 2018, 297, 117–123. [Google Scholar] [CrossRef] [PubMed]
- Guan, R.; Wang, X.; Zhao, X.; Song, N.; Zhu, J.; Wang, J.; Wang, J.; Xia, C.; Chen, Y.; Zhu, D.; et al. Emodin ameliorates bleomycin-induced pulmonary fibrosis in rats by suppressing epithelial-mesenchymal transition and fibroblast activation. Sci. Rep. 2016, 6, 35696. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Pi, Z.; Xiao, Y.; Zeng, Z.; Yu, J.; Zou, P.; Tang, B.; Qiu, X.; Tang, R.; Shi, Y.; et al. Esomeprazole alleviates fibrosis in systemic sclerosis by modulating AhR/Smad2/3 signaling. Pharmacol. Res. 2022, 176, 106057. [Google Scholar] [CrossRef]
- Abdulla, O.A.; Neamah, W.; Sultan, M.; Chatterjee, S.; Singh, N.; Nagarkatti, M.; Nagarkatti, P. AhR Ligands Differentially Regulate miRNA-132 Which Targets HMGB1 and to Control the Differentiation of Tregs and Th-17 Cells during Delayed-Type Hypersensitivity Response. Front. Immunol. 2021, 12, 635903. [Google Scholar] [CrossRef]
- Shivanna, B.; Chu, C.; Moorthy, B. The Aryl Hydrocarbon Receptor (AHR): A Novel Therapeutic Target for Pulmonary Diseases? Int. J. Mol. Sci. 2022, 23, 1516. [Google Scholar] [CrossRef]
- Takei, H.; Yasuoka, H.; Yoshimoto, K.; Takeuchi, T. Aryl hydrocarbon receptor signals attenuate lung fibrosis in the bleomycin-induced mouse model for pulmonary fibrosis through increase of regulatory T cells. Arthritis Res. Ther. 2020, 22, 20. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Wang, Y.; Zhou, Y. Beneficial roles of the AhR ligand FICZ on the regenerative potentials of BMSCs and primed cartilage templates. RSC Adv. 2022, 12, 11505–11516. [Google Scholar] [CrossRef] [PubMed]
- Fangjun, L.; Zhijia, Y. Tumor suppressive roles of eugenol in human lung cancer cells. Thorac. Cancer 2018, 9, 25–29. [Google Scholar] [CrossRef]
- Martinez-Orozco, R.; Navarro-Tito, N.; Soto-Guzman, A.; Castro-Sanchez, L.; Perez Salazar, E. Arachidonic acid promotes epithelial-to-mesenchymal-like transition in mammary epithelial cells MCF10A. Eur. J. Cell Biol. 2010, 89, 476–488. [Google Scholar] [CrossRef]
- Jin, Y.; Qi, G.; Shou, Y.; Li, D.; Liu, Y.; Guan, H.; Zhang, Q.; Chen, S.; Luo, J.; Xu, L.; et al. High throughput data-based, toxicity pathway-oriented development of a quantitative adverse outcome pathway network linking AHR activation to lung damages. J. Hazard. Mater. 2022, 425, 128041. [Google Scholar] [CrossRef] [PubMed]
- Xiao, T.; Zou, Z.; Xue, J.; Syed, B.M.; Sun, J.; Dai, X.; Shi, M.; Li, J.; Wei, S.; Tang, H.; et al. LncRNA H19-mediated M2 polarization of macrophages promotes myofibroblast differentiation in pulmonary fibrosis induced by arsenic exposure. Environ. Pollut. 2021, 268 (Pt A), 115810. [Google Scholar] [CrossRef]
- Porter, K.M.; Epstein, D.L.; Liton, P.B. Up-Regulated Expression of Extracellular Matrix Remodeling Genes in Phagocytically Challenged Trabecular Meshwork Cells. PLoS ONE 2012, 7, e34792. [Google Scholar] [CrossRef] [Green Version]
- Giangreco, A.; Lu, L.; Vickers, C.; Teixeira, V.H.; Groot, K.R.; Butler, C.R.; Ilieva, E.V.; George, P.J.; Nicholson, A.G.; Sage, E.K.; et al. β-Catenin determines upper airway progenitor cell fate and preinvasive squamous lung cancer progression by modulating epithelial–mesenchymal transition. J. Pathol. 2012, 226, 575–587. [Google Scholar] [CrossRef] [PubMed]
- Vondráček, J.; Pěnčíková, K.; Neča, J.; Ciganek, M.; Grycová, A.; Dvořák, Z.; Machala, M. Assessment of the aryl hydrocarbon receptor-mediated activities of polycyclic aromatic hydrocarbons in a human cell-based reporter gene assay. Environ. Pollut. 2017, 220, 307–316. [Google Scholar] [CrossRef] [PubMed]
- Låg, M.; Øvrevik, J.; Refsnes, M.; Holme, J.A. Potential role of polycyclic aromatic hydrocarbons in air pollution-induced non-malignant respiratory diseases. Respir. Res. 2020, 21, 299. [Google Scholar] [CrossRef]
- Go, R.; Lee, S.; Shin, Y.; Kim, M.; Park, C.; Ko, E.; Kim, S.; Choi, K. Time-dependent effect of inhaled cigarette smoke exposure in the bleomycin-induced lung injury rat model. Environ. Toxicol. 2022, 37, 1231–1243. [Google Scholar] [CrossRef]
- Hussain, M.; Xu, C.; Lu, M.; Wu, X.; Tang, L.; Wu, X. Wnt/β-catenin signaling links embryonic lung development and asthmatic airway remodeling. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 3226–3242. [Google Scholar] [CrossRef]
- Liu, J.; Xiao, Q.; Xiao, J.; Niu, C.; Li, Y.; Zhang, X.; Zhou, Z.; Shu, G.; Yin, G. Wnt/β-catenin signalling: Function, biological mechanisms, and therapeutic opportunities. Signal Transduct. Target. Ther. 2022, 7, 3. [Google Scholar] [CrossRef]
- Sun, Z.; Yang, Z.; Wang, M.; Huang, C.; Ren, Y.; Zhang, W.; Gao, F.; Cao, L.; Li, L.; Nie, S. Paraquat induces pulmonary fibrosis through Wnt/β-catenin signaling pathway and myofibroblast differentiation. Toxicol. Lett. 2020, 333, 170–183. [Google Scholar] [CrossRef]
- Gurczynski, S.J.; Pereira, N.L.; Hrycaj, S.M.; Wilke, C.; Zemans, R.L.; Moore, B.B. Stem cell transplantation uncovers TDO-AHR regulation of lung dendritic cells in herpesvirus-induced pathology. JCI Insight 2021, 6, 139965. [Google Scholar] [CrossRef] [PubMed]
- Maguire, M.; Larsen, M.C.; Foong, Y.H.; Tanumihardjo, S.; Jefcoate, C.R. Cyp1b1 deletion and retinol deficiency coordinately suppress mouse liver lipogenic genes and hepcidin expression during post-natal development. Mol. Cell. Endocrinol. 2017, 454, 50–68. [Google Scholar] [CrossRef]
- Mao, M.; Wu, Z.; Chen, J. MicroRNA-187-5p suppresses cancer cell progression in non-small cell lung cancer (NSCLC) through down-regulation of CYP1B1. Biochem. Biophys. Res. Commun. 2016, 478, 649–655. [Google Scholar] [CrossRef]
- Szychowski, K.A.; Wnuk, A.; Kajta, M.; Wójtowicz, A.K. Triclosan activates aryl hydrocarbon receptor (AhR)-dependent apoptosis and affects Cyp1a1 and Cyp1b1 expression in mouse neocortical neurons. Environ. Res. 2016, 151, 106–114. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Selvam, P.; Cheng, C.-M.; Dahms, H.-U.; Ponnusamy, V.K.; Sun, Y.-Y. AhR Mediated Activation of Pro-Inflammatory Response of RAW 264.7 Cells Modulate the Epithelial-Mesenchymal Transition. Toxics 2022, 10, 642. https://doi.org/10.3390/toxics10110642
Selvam P, Cheng C-M, Dahms H-U, Ponnusamy VK, Sun Y-Y. AhR Mediated Activation of Pro-Inflammatory Response of RAW 264.7 Cells Modulate the Epithelial-Mesenchymal Transition. Toxics. 2022; 10(11):642. https://doi.org/10.3390/toxics10110642
Chicago/Turabian StyleSelvam, Padhmavathi, Chih-Mei Cheng, Hans-Uwe Dahms, Vinoth Kumar Ponnusamy, and Yu-Yo Sun. 2022. "AhR Mediated Activation of Pro-Inflammatory Response of RAW 264.7 Cells Modulate the Epithelial-Mesenchymal Transition" Toxics 10, no. 11: 642. https://doi.org/10.3390/toxics10110642