
Metabolome and Transcriptome Association Analysis Reveals Mechanism of Synthesis of Nutrient Composition in Quinoa (Chenopodium quinoa Willd.) Seeds
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Materials
2.2. Determination of Nutritional Components of Quinoa Seeds with Four Different Colors
2.3. Extensive Targeted Metabonomic Detection and Analysis
2.4. Transcriptome Data Analysis
2.5. qRT-PCR Analysis
3. Results
3.1. Analysis of Nutritional Components of Quinoa Seeds with Different Colors
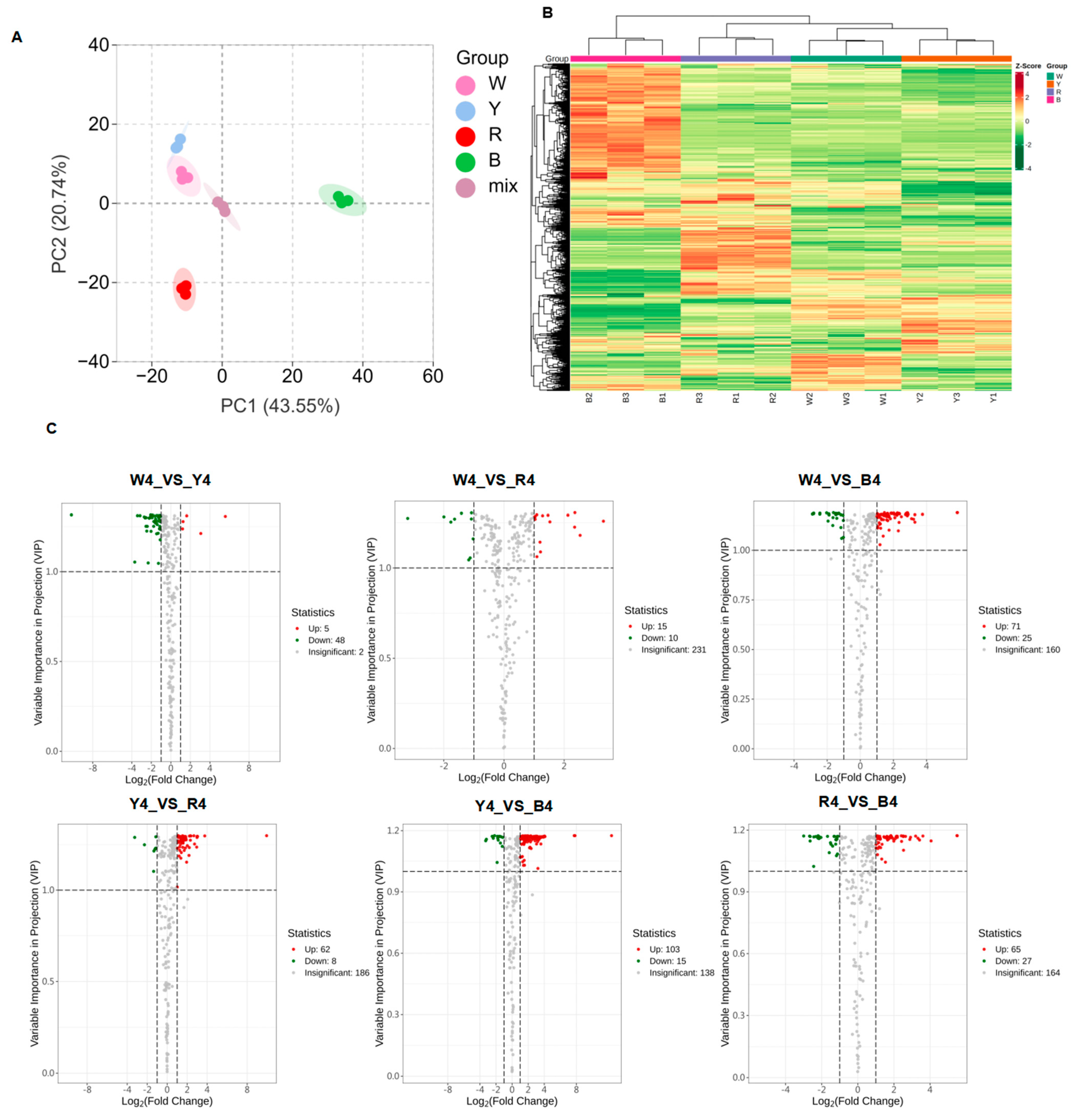
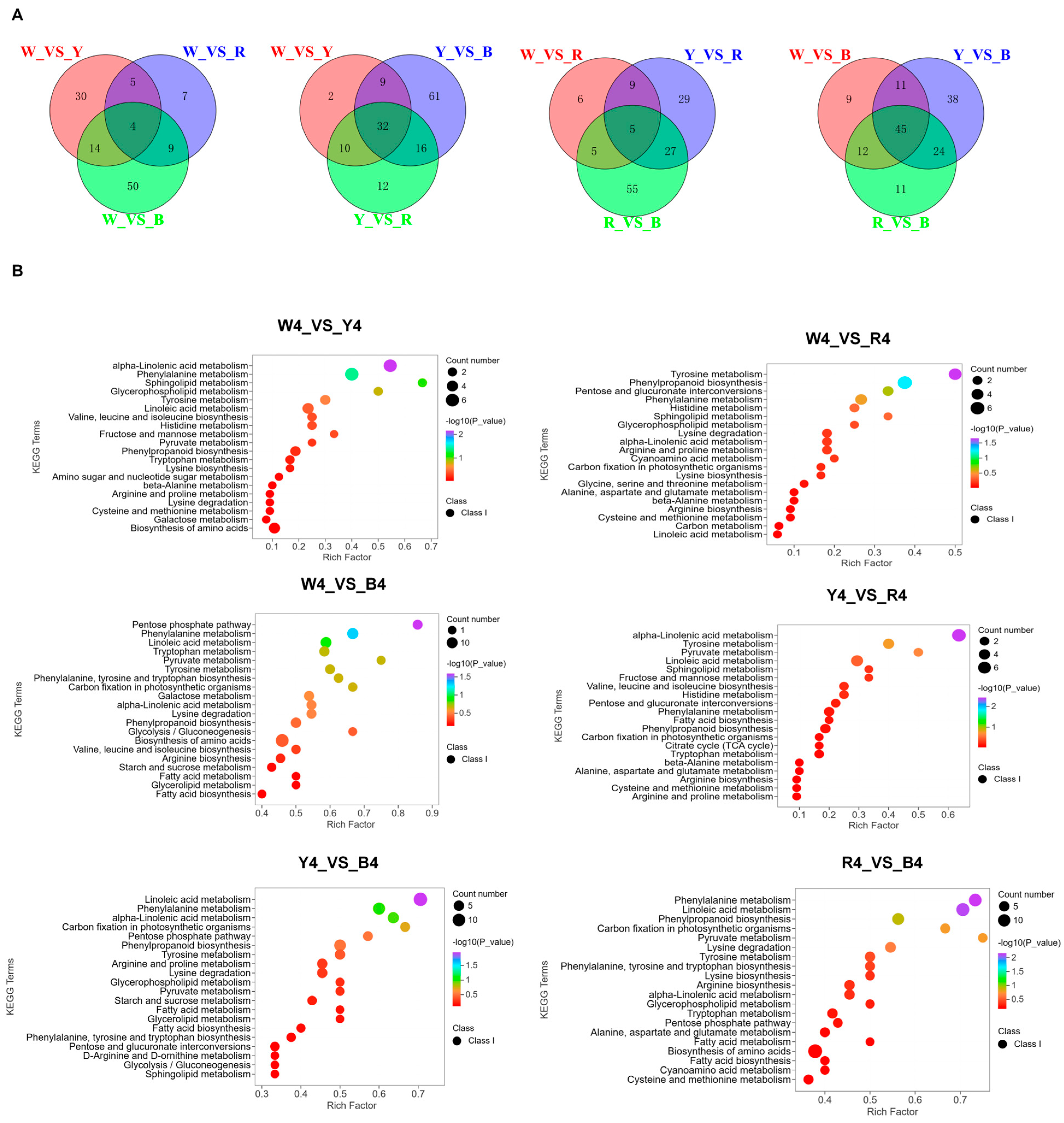
3.2. Analysis of Differential Metabolites in Quinoa Seeds with Four Different Colors
3.3. Comparative Transcriptome Analysis of Quinoa Genotypes
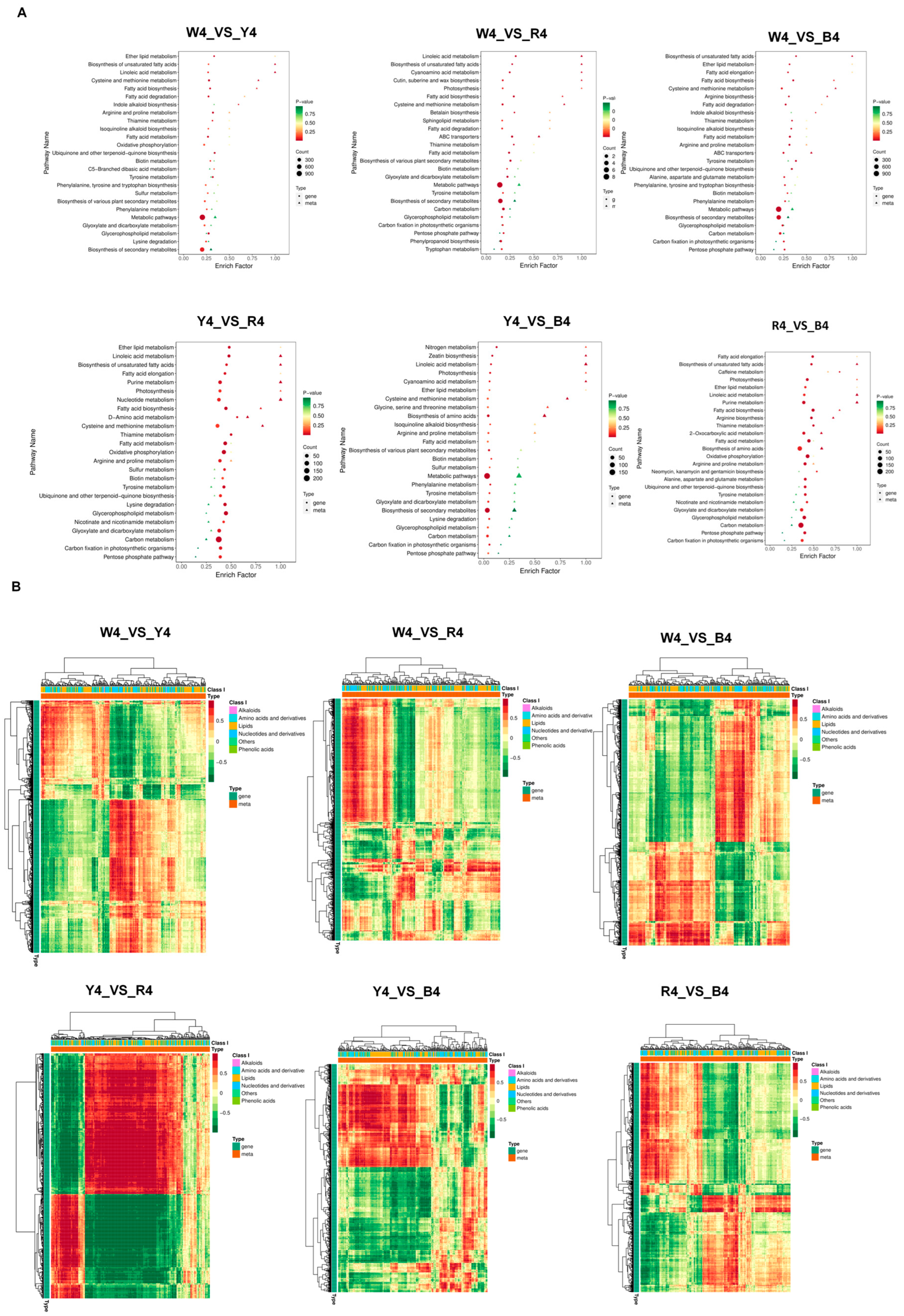
3.4. Correlation Analysis of Transcriptome and Metabolome Data
3.5. Differential Expression of Carbon Metabolism and Regulatory Genes in Four Quinoa Cultivars
3.6. Differential Expression of Lipid Anabolism and Regulatory Genes in Four Quinoa Cultivars
3.7. Differential Expression of Amino Acid Biosynthesis and Regulatory Genes in Four Quinoa Cultivars
3.8. Quantitative Reverse Transcription Polymerase Chain Reaction (qRT-PCR)
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pathan, S.; Siddiqui, R.A. Nutritional Composition and Bioactive Components in Quinoa (Chenopodium quinoa Willd.) Greens: A Review. Nutrients 2022, 14, 558. [Google Scholar] [CrossRef] [PubMed]
- Abugoch-James, L.E. Quinoa (Chenopodium quinoa Willd.): Composition, chemistry, nutritional, and functional properties. Adv. Food Nutr. Res. 2009, 58, 1–31. [Google Scholar] [PubMed]
- Villacrés, E.; Quelal, M.; Galarza, S.; Iza, D.; Silva, E. Nutritional Value and Bioactive Compounds of Leaves and Grains from Quinoa (Chenopodium quinoa Willd.). Plants 2022, 11, 213. [Google Scholar] [CrossRef] [PubMed]
- Rana, G.K.; Singh, N.K.; Deshmukh, K.K.; Mishra, S.K. Quinoa: New Light on An Old Super food-A Review. Agric. Rev. 2019, 40, 319–323. [Google Scholar]
- Burrieza, H.P.; López-Fernández, M.P.; Maldonado, S. Analogous reserve distribution and tissue characteristics in quinoa and grass seeds suggest convergent evolution. Front. Plant Sci. 2014, 5, 546. [Google Scholar] [CrossRef] [PubMed]
- Zurita-Silva, A.; Fuentes, F.; Zamora, P.; Jacobsen, S.; Schwember, A.R. Breeding quinoa (Chenopodium quinoa Willd.): Potential and perspectives. Mol. Breed. 2014, 34, 13–30. [Google Scholar] [CrossRef]
- Mu, H.; Xue, S.; Sun, Q.; Shi, J.; Zhang, D.; Wang, D.; Wei, J. Research Progress of Quinoa Seeds (Chenopodium quinoa Wild.): Nutritional Components, Technological Treatment, and Application. Foods 2023, 12, 2087. [Google Scholar] [CrossRef]
- Rizzo, A.J.; Palacios, M.B.; Vale, E.M.; Zelada, A.M.; Silveira, V.; Burrieza, H.P. Snapshot of four mature quinoa (Chenopodium quinoa) seeds: A shotgun proteomics analysis with emphasis on seed maturation, reserves and early germination. Physiol. Mol. Biol. Plants 2023, 29, 319–334. [Google Scholar] [CrossRef] [PubMed]
- Craine, E.B.; Murphy, K.M. Seed Composition and Amino Acid Profiles for Quinoa Grown in Washington State. Front. Nutr. 2020, 7, 126. [Google Scholar] [CrossRef]
- Dakhili, S.; Abdolalizadeh, L.; Hosseini, S.M.; Shojaee-Aliabadi, S.; Mirmoghtadaie, L. Quinoa protein: Composition, structure and functional properties. Food Chem. 2019, 299, 125161. [Google Scholar] [CrossRef]
- Vilcacundo, R.; Hernández-Ledesma, B. Nutritional and biological value of quinoa (Chenopodium quinoa Willd.). Curr. Opin. Food Sci. 2017, 14, 1–6. [Google Scholar] [CrossRef]
- Ren, G.X.; Teng, C.; Fan, X.; Guo, S.Y.; Zhao, G.Z.; Li, Z.; Liang, Z.; Qin, P.Y. Nutrient composition, functional activity and industrial applications of quinoa (Chenopodium quinoa Willd.). Food Chem. 2023, 410, 135290. [Google Scholar] [CrossRef] [PubMed]
- Xie, H.; Zhang, P.; Jiang, C.; Wang, Q.; Guo, Y.; Zhang, X.; Huang, T.; Liu, J.; Li, L.; Li, H.; et al. Combined transcriptomic and metabolomic analyses of high temperature stress response of quinoa seedlings. BMC Plant Biol. 2023, 23, 292. [Google Scholar] [CrossRef]
- Niu, M.; Chen, X.; Zhou, W.; Guo, Y.; Yuan, X.; Cui, J.; Shen, Z.; Su, N. Multi-omics analysis provides insights intro lysine accumulation in quinoa (Chenopodium quinoa Willd.) sprouts. Food Res. Int. 2023, 171, 113026. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Ma, Y.; Li, J.; Liu, B.; Liu, X.; Zhang, J.; Zhang, M.; Wang, C.; Zhang, L.; Lv, W.; et al. Transcriptomics-metabolomics joint analysis: New highlight into the triterpenoid saponin biosynthesis in quinoa (Chenopodium quinoa Willd.). Front. Plant Sci. 2022, 13, 964558. [Google Scholar] [CrossRef]
- Hao, Y.; Hong, Y.; Guo, H.; Qin, P.; Huang, A.; Yang, X.; Ren, G. Transcriptomic and metabolomic landscape of quinoa during seed germination. BMC Plant Biol. 2022, 22, 237. [Google Scholar] [CrossRef] [PubMed]
- Kjeldahl, J. Neue Methode zur Bestimmung des Stickstoffs in organischen Körpern. Fresenius’ J. Anal. Chem. 1883, 22, 366–382. [Google Scholar] [CrossRef]
- Hewavitharana, G.G.; Perera, D.N.; Navaratne, S.B.; Wickramasinghe, I. Extraction methods of fat from food samples and preparation of fatty acid methyl esters for gas chromatography: A review. Arab. J. Chem. 2020, 13, 6865–6875. [Google Scholar] [CrossRef]
- Chen, Y.; Xie, M.Y.; Nie, S.P.; Li, C.; Wang, Y.X. Composition analysis and antioxidant activity of a polysaccharide from the fruiting bodies of Ganoderma atrum. Food Chem. 2008, 107, 231–241. [Google Scholar] [CrossRef]
- Li, L.; Zhang, H.; Liu, J.; Huang, T.; Zhang, X.; Xie, H.; Guo, Y.; Wang, Q.; Zhang, P.; Qin, P. Grain color formation and analysis of correlated genes by metabolome and transcriptome in different wheat lines at maturity. Front. Nutr. 2023, 10, 1112497. [Google Scholar] [CrossRef]
- Ma, D.; Xu, B.; Feng, J.; Hu, H.; Tang, J.; Yin, G.; Xie, Y.; Wang, C. Dynamic metabolomics and transcriptomics analyses for characterization of phenolic compounds and their biosynthetic characteristics in wheat grain. Front. Nutr. 2022, 9, 844337. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Zhang, X.; Hou, H.; Ma, X.; Sun, S.; Wang, H.; Kong, L. Metabolomics and gene expression analysis reveal the accumulation patterns of phenylpropanoids and flavonoids in different colored-grain wheats (Triticum aestivum L.). Food Res. Int. 2020, 138, 109711. [Google Scholar] [CrossRef] [PubMed]
- Knappe, S.; Flugge, U.I.; Fischer, K. Analysis of the Plastidic phosphate translocator Gene Family in Arabidopsis and Identification of New phosphate translocator-Homologous Transporters, Classified by Their Putative Substrate-Binding Site. Plant Physiol. 2003, 131, 1178–1190. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for Rna-Seq data with Deseq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Wei, X.; Liu, Z.; Wu, X.; Bao, C.; Sun, Y.; Su, N.; Cui, J. Transcriptome Analysis Reveals the Molecular Mechanism of GABA Accumulation during Quinoa (Chenopodium quinoa Willd.) Germination. J. Agric. Food Chem. 2021, 69, 12171–12186. [Google Scholar] [CrossRef] [PubMed]
- Izumi, M.; Tsunoda, H.; Suzuki, Y.; Makino, A.; Ishida, H. RBCS1A and RBCS3B, two major members within the Arabidopsis RBCS multigene family, function to yield sufficient Rubisco content for leaf photosynthetic capacity. J. Exp. Bot. 2013, 63, 2159–2170. [Google Scholar] [CrossRef] [PubMed]
- Kleczkowski, L.A. Glucose activation and metabolism through UDP-glucose pyrophosphorylase in plants. Phytochemistry 1994, 37, 1507–1515. [Google Scholar] [CrossRef]
- Kleczkowski, L.A.; Geisler, M.; Ciereszko, I.; Johansson, H. UDP-glucose pyrophosphorylase. An old protein with new tricks. Plant Physiol. 2004, 134, 912–918. [Google Scholar] [CrossRef]
- Malinova, I.; Kunz, H.H.; Alseekh, S.; Herbst, K.; Fernie, A.R.; Gierth, M.; Fettke, J. Reduction of the cytosolic phosphoglucomutase in Arabidopsis reveals impact on plant growth, seed and root development, and carbohydrate partitioning. PLoS ONE 2014, 9, 112468. [Google Scholar] [CrossRef]
- Liao, Y.Q.; Ji, D.H.; Xu, K.; Chen, C.S.; Wang, W.L.; Xie, C.T. Cloning and functional analysis of a phosphoglycerate kinase (PhPGK) from Pyropia haitanensis. J. Appl. Phycol. 2023, 35, 1933–1943. [Google Scholar] [CrossRef]
- Rosa-Téllez, S.; Anoman, A.D.; Flores-Tornero, M.; Toujani, W.; Alseek, S.; Fernie, A.R.; Nebauer, S.G.; Muñoz-Bertomeu, J.; Segura, J.; Ros, R. Phosphoglycerate Kinases Are Co-Regulated to Adjust Metabolism and to Optimize Growth. Plant Physiol. 2018, 176, 1182–1198. [Google Scholar] [CrossRef] [PubMed]
- Bi, H.G.; Wang, M.L.; Dong, X.B.; Ai, X.Z. Cloning and expression analysis of transketolase gene in Cucumis sativus L. Plant Physiol. Biochem. 2013, 70, 512–521. [Google Scholar] [CrossRef] [PubMed]
- Bi, H.G.; Wang, M.L.; Dong, X.B.; Ai, X.Z. Decreased TK activity alters growth, yield and tolerance to low temperature and low light intensity in transgenic cucumber plants. Plant Cell Rep. 2015, 34, 345–354. [Google Scholar] [CrossRef] [PubMed]
- Cai, Y.C.; Li, S.F.; Jiao, G.A.; Sheng, Z.H.; Wu, Y.W.; Shao, G.N.; Xie, L.H.; Peng, C.; Xu, J.F.; Tang, S.P.; et al. OsPK2 encodes a plastidic pyruvate kinase involved in rice endosperm starch synthesis, compound granule formation and grain filling. Plant Biotechnol. J. 2018, 16, 1878–1891. [Google Scholar] [CrossRef] [PubMed]
- Streb, S.; Zeeman, S.C. Starch metabolism in Arabidopsis. Arab. Book 2012, 10, e0160. [Google Scholar] [CrossRef] [PubMed]
- Gautam, T.; Dutta, M.; Jaiswal, V.; Zinta, G.; Gahlaut, V.; Kumar, S. Emerging Roles of SWEET Sugar Transporters in Plant Development and Abiotic Stress Responses. Cells 2022, 11, 1303. [Google Scholar] [CrossRef] [PubMed]
- Qu, A.L.; Xu, Y.; Yu, X.X.; Si, Q.; Xu, X.W.; Liu, C.H.; Yang, L.Y.; Zheng, Y.P.; Zhang, M.M.; Zhang, S.Q.; et al. Sporophytic control of anther development and male fertility by glucose-6-phosphate/phosphate translocator 1 (OsGPT1) in rice. J. Genet. Genom. 2021, 48, 695–705. [Google Scholar] [CrossRef] [PubMed]
- Li-Beisson, Y.; Shorrosh, B.; Beisson, F.; Andersson, M.X.; Arondel, V.; Bates, P.D.; Baud, S.; Bird, D.; DeBono, A.; Durrett, T.P.; et al. Acyl-Lipid Metabolism. Arab. B 2013, 11, e0161. [Google Scholar] [CrossRef] [PubMed]
- Shen, G.D.; Sun, W.L.; Chen, Z.C.; Shi, L.; Hong, J.; Shi, J.X. Plant GDSL Esterases/Lipases: Evolutionary, Physiological and Molecular Functions in Plant Development. Plants 2022, 11, 468. [Google Scholar] [CrossRef]
- Zhao, L.; Haslam, T.M.; Sonntag, A.; Molina, I.; Kunst, L. Functional Overlap of Long-Chain Acyl-CoA Synthetases in Arabidopsis. Plant Cell Physiol. 2019, 60, 1041–1054. [Google Scholar] [CrossRef]
- Fan, K.; Qin, Y.; Hu, X.; Xu, J.; Ye, Q.; Zhang, C.; Ding, Y.; Li, G.; Chen, Y.; Liu, J.; et al. Identification of genes associated with fatty acid biosynthesis based on 214 safflower core germplasm. BMC Genom. 2023, 24, 763. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Loh, K.; Song, Z.Y.; Yang, H.Q.; Zhang, Y.; Lin, S. Update on glycerol-3-phosphate acyltransferases: The roles in the development of insulin resistance. Nutr. Diabetes 2018, 8, 34. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.L.; Huang, X.S.; Hu, T.; Chen, S.W.; Wang, Y.; Shi, X.F.; Yin, M.; Li, R.Z.; Wang, J.P.; Jia, X.J. Genome-Wide Analysis of Glycerol-3-Phosphate Acyltransferase (GPAT) Family in Perilla frutescensand Functional Characterization of PfGPAT9 Crucial for Biosynthesis of Storage Oils Rich in High-Value Lipids. Int. J. Mol. Sci. 2023, 24, 15106. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.; Liu, X.; Yin, X.M.; Dong, T.; Yu, M.; Wu, Y. Rice Non-Specific Phospholipase C6 Is Involved in Mesocotyl Elongation. Plant Cell Physiol. 2021, 62, 985–1000. [Google Scholar] [CrossRef] [PubMed]
- Tremblay, J.; Goulet, M.C.; Michaud, D. Recombinant cystatins in plants. Biochimie 2019, 166, 184–193. [Google Scholar] [CrossRef] [PubMed]
- Pegg, A.E. S-Adenosylmethionine decarboxylase. Essays Biochem. 2009, 46, 25–45. [Google Scholar] [PubMed]
- Sekula, B.; Ruszkowski, M.; Dauter, Z. S-adenosylmethionine synthases in plants: Structural characterization of type I and II isoenzymes from Arabidopsis thaliana and Medicago truncatula. Int. J. Biol. Macromol. 2020, 151, 554–565. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Tang, L.; Qiu, J.; Zhang, W.; Wang, Y.; Tong, X.; Wei, X.; Hou, Y.; Zhang, J. Serine carboxypeptidase 46 Regulates Grain Filling and Seed Germination in Rice (Oryza sativa L.). PLoS ONE 2016, 11, e0159737. [Google Scholar] [CrossRef]
- Suzuki, A.; Knaff, D.B. Glutamate synthase: Structural, mechanistic and regulatory properties, and role in the amino acid metabolism. Photosynth. Res. 2005, 83, 191–217. [Google Scholar] [CrossRef]
- Walker, M.C.; Donk, W.A. The many roles of glutamate in metabolism. J. Ind. Microbiol. Biotechnol. 2016, 43, 419–430. [Google Scholar] [CrossRef]
- Marino, D.; Cañas, R.A.; Betti, M. Is plastidic glutamine synthetase essential for C plants? A tale of photorespiratory mutants, ammonium tolerance and conifers. New Phytol. 2022, 234, 1559–1565. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.-T.; Liao, H.-S.; Hsieh, M.-H. Glutamine Metabolism, Sensing and Signaling in Plants. Plant Cell Physiol. 2023, 64, 1466–1481. [Google Scholar] [CrossRef] [PubMed]
- Guo, Q.; Chen, X.; Li, B. Purification and characterization of tomato arginine decarboxylase and its inhibition by the bacterial small molecule phevamine A. Protein Expr. Purif. 2023, 210, 106326. [Google Scholar] [CrossRef] [PubMed]
- Yin, R.; Frey, M.; Gierl, A.; Glawischnig, E. Plants contain two distinct classes of functional tryptophan synthase beta proteins. Phytochemistry 2010, 71, 1667–1672. [Google Scholar] [CrossRef] [PubMed]
- Claudie, R.; Luis, O.E.; Jean-Bernard, C.; Anis, M. Limami, Characterization of alanine aminotransferase (AlaAT) multigene family and hypoxic response in young seedlings of the model legume Medicago truncatula. J. Exp. Bot. 2006, 57, 3079–3089. [Google Scholar]
- Hundertmark, M.; Hincha, D.K. LEA (Late Embryogenesis Abundant) proteins and their encoding genes in Arabidopsis thaliana. BMC Genom. 2008, 9, 118. [Google Scholar] [CrossRef]
- Manfre, A.J.; Lanni, L.M.; Marcotte, W.R., Jr. The Arabidopsis group 1 LATE EMBRYOGENESIS ABUNDANT protein ATEM6 is required for normal seed development. Plant Physiol. 2006, 140, 140–149. [Google Scholar] [CrossRef] [PubMed]
- Edstam, M.M.; Viitanen, L.; Salminen, T.A.; Edqvist, J. Evolutionary history of the non-specific lipid transfer proteins. Mol. Plant 2011, 4, 947–964. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Wang, L.; Tang, B.; Ren, R.; Shi, T.; Zhu, L.; Deng, J.; Liang, C.; Wang, Y.; Chen, Q. Integrated Transcriptomics and Widely Targeted Metabolomics Analyses Provide Insights Into Flavonoid Biosynthesis in the Rhizomes of Golden Buckwheat (Fagopyrum cymosum). Front. Plant Sci. 2022, 13, 803472. [Google Scholar] [CrossRef]
- Pedrali, D.; Giupponi, L.; De la Peña-Armada, R.; Villanueva-Suárez, M.J.; Mateos-Aparicio, I. The quinoa variety influences the nutritional and antioxidant profile rather than the geographic factors. Food Chem. 2023, 402, 133531. [Google Scholar] [CrossRef]
- Feng, Y.; Fan, X.; Zhang, S.; Wu, T.; Bai, L.; Wang, H.; Ma, Y.; Guan, X.; Wang, C.; Yang, H. Effects of variety and origin on the metabolic and texture characteristics of quinoa seeds based on ultrahigh-performance liquid chromatography coupled with high-field quadrupole-orbitrap high-resolution mass spectrometry. Food Res. Int. 2022, 162, 111693. [Google Scholar] [CrossRef] [PubMed]
- Gargiulo, L.; Grimberg, Å.; Repo-Carrasco-Valencia, R.; Carlsson, A.S.; Mele, G. Morpho-densitometric traits for quinoa (Chenopodium quinoa Willd.) seed phenotyping by two X-ray micro-CT scanning approaches. J. Cereal Sci. 2019, 90, 102829. [Google Scholar] [CrossRef]
- Baud, S.; Boutin, J.P.; Miquel, M.; Lepiniec, L.; Rochat, C. An integrated overview of seed development in Arabidopsis thaliana ecotype WS. Plant Physiol. Biochem. 2002, 40, 151–160. [Google Scholar] [CrossRef]
- Zhang, C.; Zhang, P.; Zhang, X.; Wang, Q.; Liu, J.; Li, L.; Cheng, S.; Qin, P. Integrated Metabolome and Transcriptome Analyses Reveal Amino Acid Biosynthesis Mechanisms during the Physiological Maturity of Grains in Yunnan Hulled Wheat (Triticum aestivum ssp. Yunnanense King). Int. J. Mol. Sci. 2023, 24, 13475. [Google Scholar] [CrossRef]
- Johnston, M.L.; Luethy, M.H.; Miernyk, J.A.; Randall, D.D. Cloning and molecular analyses of the Arabidopsis thaliana plastid pyruvate dehydrogenase subunits. Biochim. Biophys. Acta 1997, 1321, 200–206. [Google Scholar] [CrossRef]
- Andre, C.; Froehlich, J.E.; Moll, M.R.; Benning, C. A heteromeric plastidic pyruvate kinase complex involved in seed oil biosynthesis in Arabidopsis. Plant Cell 2007, 19, 2006–2022. [Google Scholar] [CrossRef] [PubMed]
- Xu, Q.; Yi, S.; Ma, Y.; Song, M.; Song, Y.; Mu, S.; Li, Y.; Liu, X.; Ren, Y.; Gao, C.; et al. Carbon export from leaves is controlled via ubiquitination and phosphorylation of sucrose transporter SUC2. Proc. Natl. Acad. Sci. USA 2020, 117, 6223–6230. [Google Scholar] [CrossRef] [PubMed]
- Cordoba, E.; Aceves-Zamudio, D.L.; Hernández-Bernal, A.F.; Ramos-Vega, M.; León, P. Sugar regulation of SUGAR TRANSPORTER PROTEIN 1 (STP1) expression in Arabidopsis thaliana. J. Exp. Bot. 2015, 66, 147–159. [Google Scholar] [CrossRef] [PubMed]
- Lin, W.H.; Ye, R.; Ma, H.; Xu, Z.H.; Xue, H.W. DNA chip-based expression profile analysis indicates involvement of the phosphatidylinositol signaling pathway in multiple plant responses to hormone and abiotic treatments. Cell Res. 2004, 14, 34–45. [Google Scholar] [CrossRef]
- Kazaz, S.; Barthole, G.; Domergue, F.; Ettaki, H.; To, A.; Vasselon, D.; De Vos, D.; Belcram, K.; Lepiniec, L.; Baud, S. Differential Activation of Partially Redundant Δ9 Stearoyl-ACP Desaturase Genes Is Critical for Omega-9 Monounsaturated Fatty Acid Biosynthesis During Seed Development in Arabidopsis. Plant Cell 2020, 32, 3613–3637. [Google Scholar] [CrossRef]
- Baud, S.; Lepiniec, L. Physiological and developmental regulation of seed oil production. Prog. Lipid Res. 2010, 49, 235–249. [Google Scholar] [CrossRef] [PubMed]
- Krčková, Z.; Brouzdová, J.; Daněk, M.; Kocourková, D.; Rainteau, D.; Ruelland, E.; Valentová, O.; Pejchar, P.; Martinec, J. Arabidopsis non-specific phospholipase C1: Characterization and its involvement in response to heat stress. Front. Plant Sci. 2015, 6, 928. [Google Scholar] [CrossRef] [PubMed]
- Gaude, N.; Nakamura, Y.; Scheible, W.R.; Ohta, H.; Dörmann, P. Phospholipase C5 (NPC5) is involved in galactolipid accumulation during phosphate limitation in leaves of Arabidopsis. Plant J. 2008, 56, 28–39. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Tan, H.; Zhang, C.; Li, Q.; Liu, Q. Starch biosynthesis in cereal endosperms: An updated review over the last decade. Plant Commun. 2021, 2, 100237. [Google Scholar] [CrossRef] [PubMed]
- Schwender, J.; Goffman, F.; Ohlrogge, J.; Yair, H. Rubisco without the Calvin cycle improves the carbon efficiency of developing green seeds. Nature 2004, 432, 779–782. [Google Scholar] [CrossRef]
- Patil, G.; Vuong, T.D.; Kale, S.; Valliyodan, B.; Deshmukh, R.; Zhu, C.; Wu, X.; Bai, Y.; Yungbluth, D.; Lu, F.; et al. Dissecting genomic hotspots underlying seed protein, oil, and sucrose content in an interspecific mapping population of soybean using high-density linkage mapping. Plant Biotechnol. J. 2018, 16, 1939–1953. [Google Scholar] [CrossRef]
- Bates, P.D.; Stymne, S.; Ohlrogge, J. Biochemical pathways in seed oil synthesis. Curr. Opin. Plant Biol. 2013, 16, 358–364. [Google Scholar] [CrossRef]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, J.; Wang, Y.; Sun, J.; Li, Y.; Zhu, R.; Yin, Y.; Wang, C.; Yin, X.; Qin, L. Metabolome and Transcriptome Association Analysis Reveals Mechanism of Synthesis of Nutrient Composition in Quinoa (Chenopodium quinoa Willd.) Seeds. Foods 2024, 13, 1325. https://doi.org/10.3390/foods13091325
Yang J, Wang Y, Sun J, Li Y, Zhu R, Yin Y, Wang C, Yin X, Qin L. Metabolome and Transcriptome Association Analysis Reveals Mechanism of Synthesis of Nutrient Composition in Quinoa (Chenopodium quinoa Willd.) Seeds. Foods. 2024; 13(9):1325. https://doi.org/10.3390/foods13091325
Chicago/Turabian StyleYang, Jindan, Yiyun Wang, Jiayi Sun, Yuzhe Li, Renbin Zhu, Yongjie Yin, Chuangyun Wang, Xuebin Yin, and Lixia Qin. 2024. "Metabolome and Transcriptome Association Analysis Reveals Mechanism of Synthesis of Nutrient Composition in Quinoa (Chenopodium quinoa Willd.) Seeds" Foods 13, no. 9: 1325. https://doi.org/10.3390/foods13091325