
Genetic-Phenotype Analysis of Bifidobacterium bifidum and Its Glycoside Hydrolase Gene Distribution at Different Age Groups
 , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Bacterial Screening
2.2. Genomic DNA Extraction
2.3. Genome Sequencing, Assembly, and Annotation
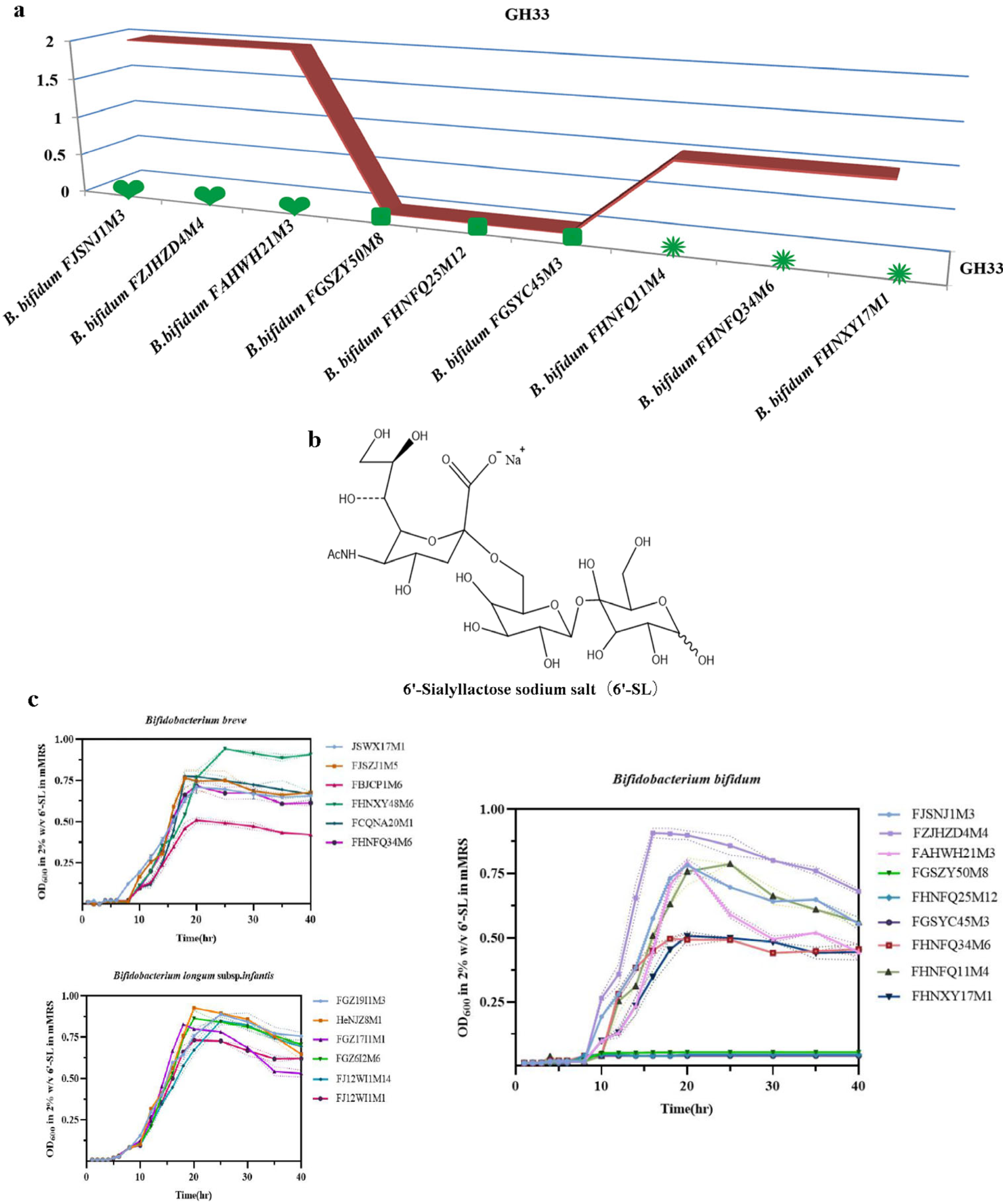
2.4. Determination of the Ability of Bifidobacteria to Utilize 6’-Sialyllactose
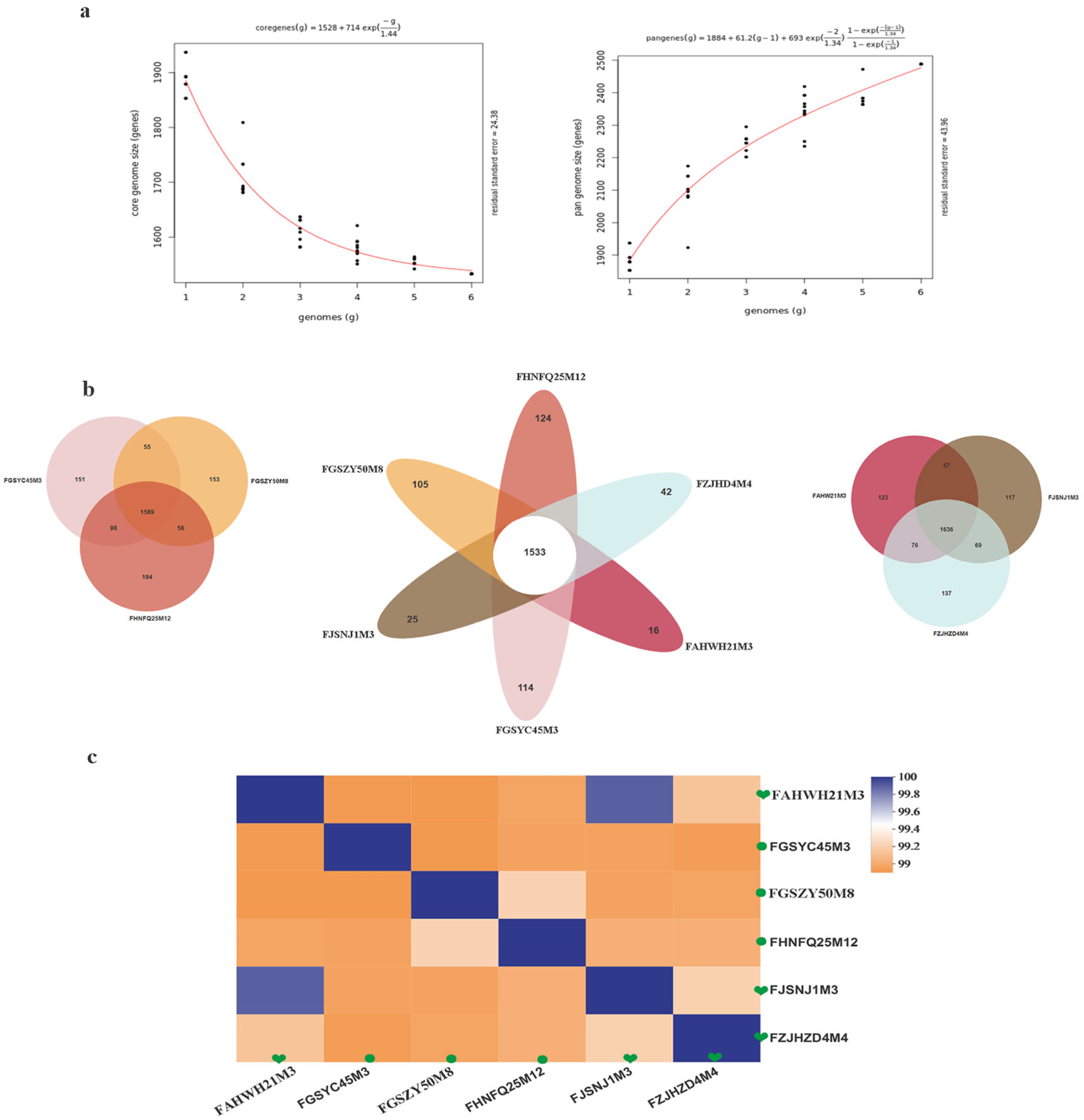
2.5. Comparative Analysis of the Genomes of Six B. bifidum Strains from Subjects of Different Ages
2.6. Antibiotic Resistance Gene and Phenotype Analysis of B. bifidum
2.7. Statistical Analysis
3. Results
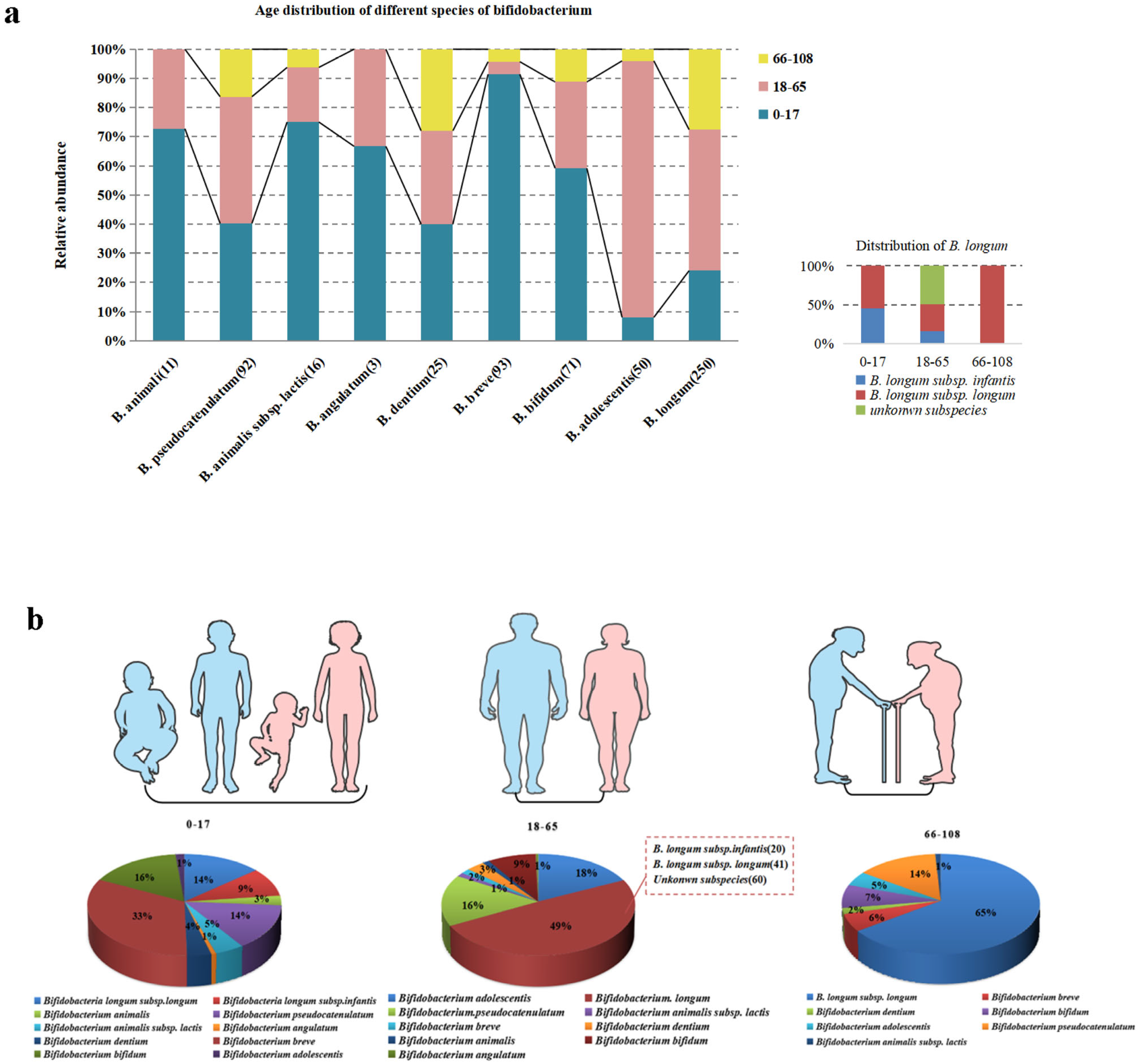
3.1. Bifidobacteria Distribution at Various Ages
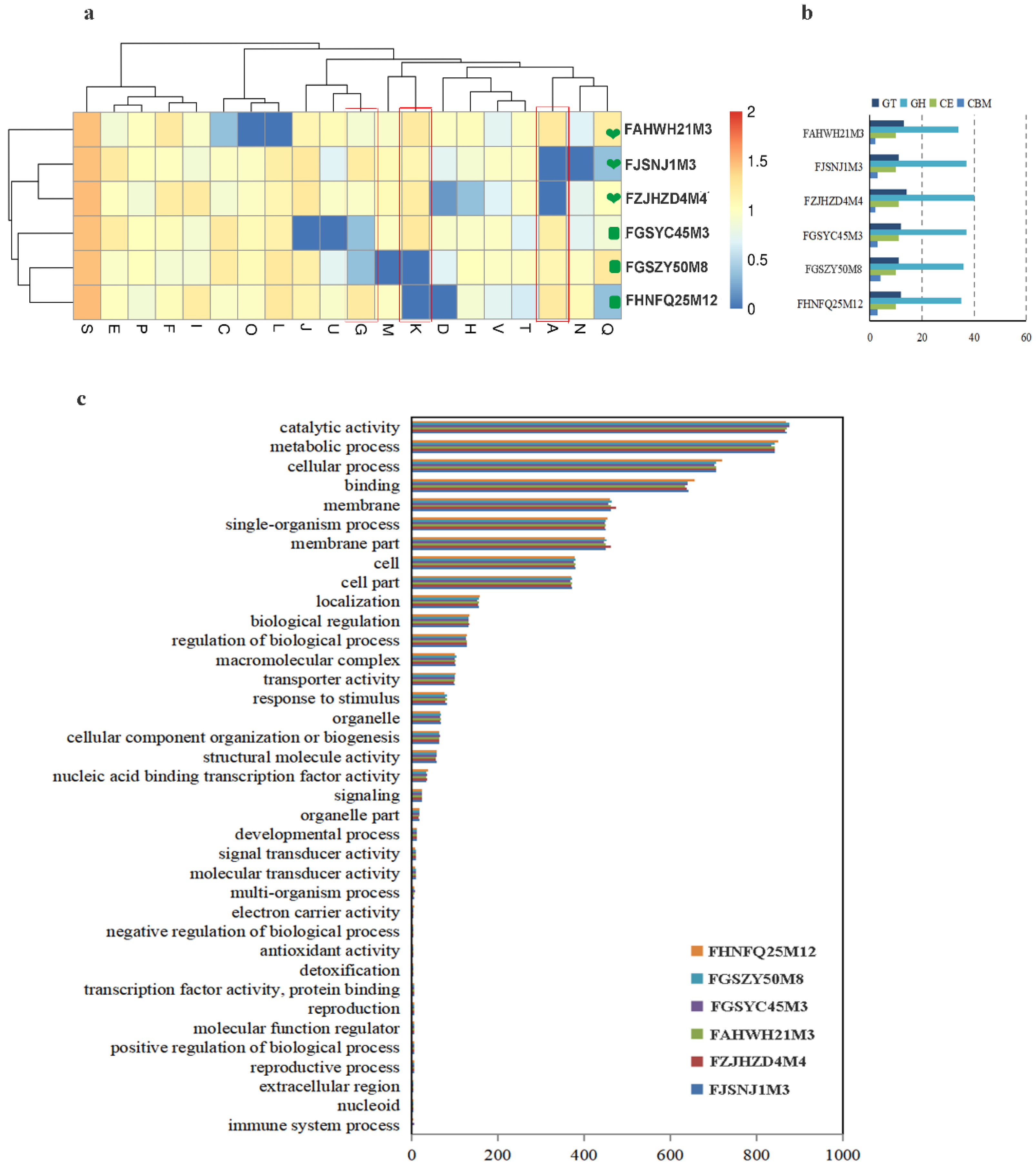
3.2. Glycoside Hydrolase Genes of Bifidobacterium Species Was Studied at Various Ages
3.3. Determination of the Ability of Bifidobacteria to Utilize 6’-Sialyllactose
3.4. Comparison of Genomes between Six Strains of B. bifidum from Subjects of Different Ages
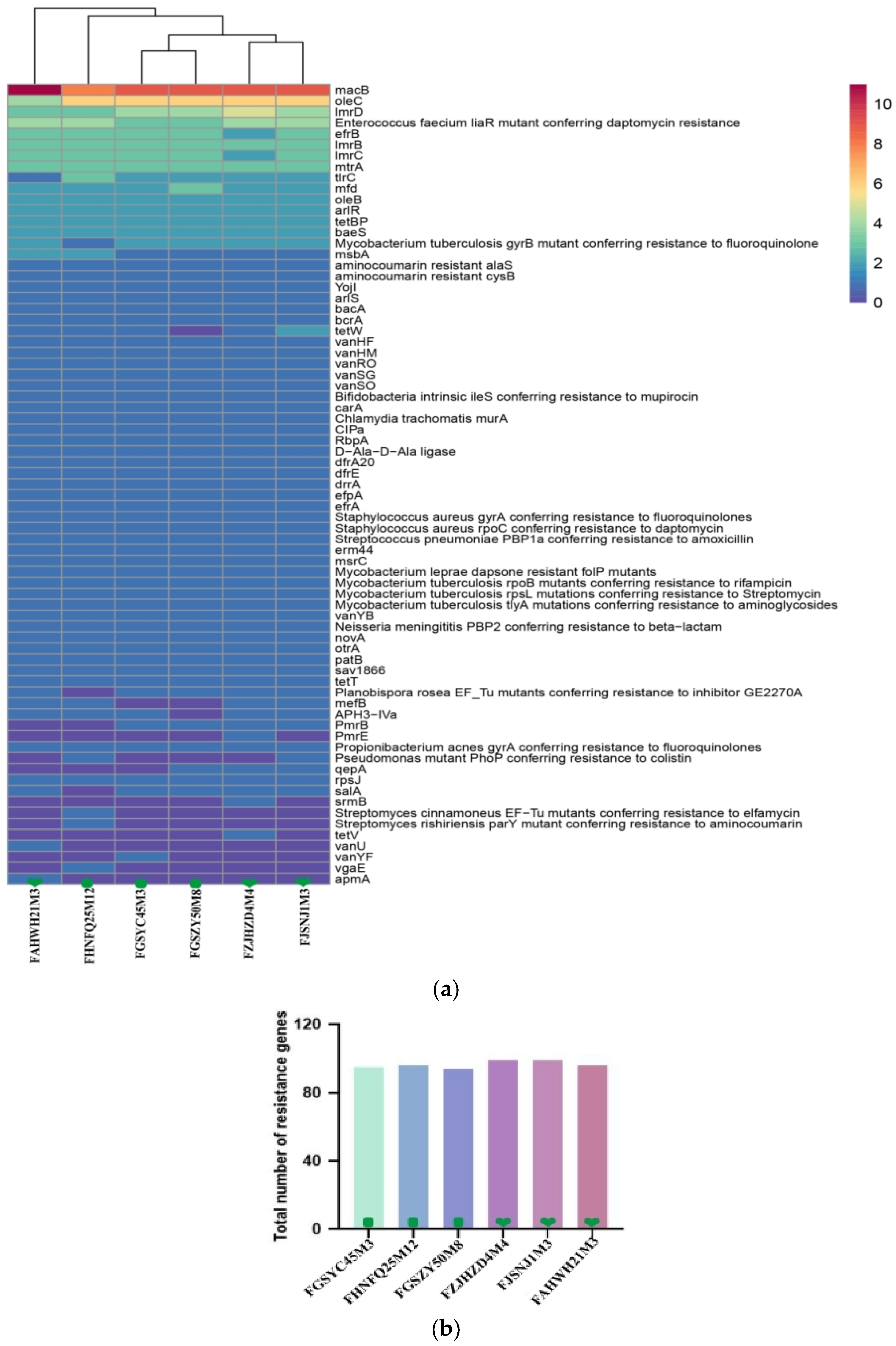
3.5. Antibiotic Resistance Gene and Phenotype Analysis in B. bifidum
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Costea, P.I.; Hildebrand, F.; Arumugam, M.; Backhed, F.; Blaser, M.J.; Bushman, F.D.; de Vos, W.M.; Ehrlich, S.D.; Fraser, C.M.; Hattori, M.; et al. Enterotypes in the Landscape of Gut Microbial Community Composition. Nat. Microbiol. 2018, 3, 388. [Google Scholar] [CrossRef] [Green Version]
- Human Microbiome Project Consortium. Structure, Function and Diversity of the Healthy Human Microbiome. Nature 2012, 486, 207–214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sender, R.; Fuchs, S.; Milo, R. Revised Estimates for the Number of Human and Bacteria Cells in the Body. PLoS Biol. 2016, 14, e1002533. [Google Scholar] [CrossRef] [Green Version]
- Yatsunenko, T.; Rey, F.E.; Manary, M.J.; Trehan, I.; Dominguez-Bello, M.G.; Contreras, M.; Magris, M.; Hidalgo, G.; Baldassano, R.N.; Anokhin, A.P.; et al. Human Gut Microbiome Viewed across Age and Geography. Nature 2012, 486, 222–227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Falony, G.; Joossens, M.; Vieira-Silva, S.; Wang, J.; Darzi, Y.; Faust, K.; Kurilshikov, A.; Bonder, M.J.; Valles-Colomer, M.; Vandeputte, D.; et al. Population-Level Analysis of Gut Microbiome Variation. Science 2016, 352, 560–564. [Google Scholar] [CrossRef]
- Setbo, E.; Campbell, K.; O’Cuiv, P.; Hubbard, R. Utility of Probiotics for Maintenance or Improvement of Health Status in Older People—A Scoping Review. J. Nutr. Health Aging 2019, 23, 364–372. [Google Scholar] [CrossRef] [PubMed]
- Walter, J.; Maldonado-Gómez, M.X.; Martínez, I. To Engraft or Not to Engraft: An Ecological Framework for Gut Microbiome Modulation with Live Microbes. Curr. Opin. Biotechnol. 2018, 49, 129–139. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Wang, L.; Guo, Z.; Sun, Z.; Gesudu, Q.; Kwok, L.; Menghebilige; Zhang, H. 454 Pyrosequencing Reveals Changes in the Faecal Microbiota of Adults Consuming Lactobacillus Casei Zhang. FEMS Microbiol. Ecol. 2014, 88, 612–622. [Google Scholar] [CrossRef] [Green Version]
- Koenig, J.E.; Spor, A.; Scalfone, N.; Fricker, A.D.; Stombaugh, J.; Knight, R.; Angenent, L.T.; Ley, R.E. Succession of Microbial Consortia in the Developing Infant Gut Microbiome. Proc. Natl. Acad. Sci. USA 2011, 108 (Suppl. 1), 4578–4585. [Google Scholar] [CrossRef] [Green Version]
- Roger, L.C.; Costabile, A.; Holland, D.T.; Hoyles, L.; McCartney, A.L. Examination of Faecal Bifidobacterium Populations in Breast- and Formula-Fed Infants during the First 18 Months of Life. Microbiology 2010, 156, 3329–3341. [Google Scholar] [CrossRef] [Green Version]
- Matsuki, T.; Yahagi, K.; Mori, H.; Matsumoto, H.; Hara, T.; Tajima, S.; Ogawa, E.; Kodama, H.; Yamamoto, K.; Yamada, T.; et al. A Key Genetic Factor for Fucosyllactose Utilization Affects Infant Gut Microbiota Development. Nat. Commun. 2016, 7, 11939. [Google Scholar] [CrossRef]
- O’Callaghan, A.; van Sinderen, D. Bifidobacteria and Their Role as Members of the Human Gut Microbiota. Front. Microbiol. 2016, 7, 925. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yassour, M.; Vatanen, T.; Siljander, H.; Hämäläinen, A.-M.; Härkönen, T.; Ryhänen, S.J.; Franzosa, E.A.; Vlamakis, H.; Huttenhower, C.; Gevers, D.; et al. Natural History of the Infant Gut Microbiome and Impact of Antibiotic Treatment on Bacterial Strain Diversity and Stability. Sci. Transl. Med. 2016, 8, 343ra81. [Google Scholar] [CrossRef] [Green Version]
- Sabbioni, A.; Ferrario, C.; Milani, C.; Mancabelli, L.; Riccardi, E.; Di Ianni, F.; Beretti, V.; Superchi, P.; Ossiprandi, M.C. Modulation of the Bifidobacterial Communities of the Dog Microbiota by Zeolite. Front. Microbiol. 2016, 7, 1491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bäckhed, F.; Roswall, J.; Peng, Y.; Feng, Q.; Jia, H.; Kovatcheva-Datchary, P.; Li, Y.; Xia, Y.; Xie, H.; Zhong, H.; et al. Dynamics and Stabilization of the Human Gut Microbiome during the First Year of Life. Cell Host Microbe 2015, 17, 690–703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turroni, F.; Foroni, E.; Pizzetti, P.; Giubellini, V.; Ribbera, A.; Merusi, P.; Cagnasso, P.; Bizzarri, B.; de’Angelis, G.L.; Shanahan, F.; et al. Exploring the Diversity of the Bifidobacterial Population in the Human Intestinal Tract. Appl. Environ. Microbiol. 2009, 75, 1534–1545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woodmansey, E.J.; McMurdo, M.E.T.; Macfarlane, G.T.; Macfarlane, S. Comparison of Compositions and Metabolic Activities of Fecal Microbiotas in Young Adults and in Antibiotic-Treated and Non-Antibiotic-Treated Elderly Subjects. Appl. Environ. Microbiol. 2004, 70, 6113–6122. [Google Scholar] [CrossRef] [Green Version]
- Turroni, F.; Milani, C.; Duranti, S.; Mahony, J.; van Sinderen, D.; Ventura, M. Glycan Utilization and Cross-Feeding Activities by Bifidobacteria. Trends Microbiol. 2018, 26, 339–350. [Google Scholar] [CrossRef]
- Turroni, F.; Bottacini, F.; Foroni, E.; Mulder, I.; Kim, J.-H.; Zomer, A.; Sánchez, B.; Bidossi, A.; Ferrarini, A.; Giubellini, V.; et al. Genome Analysis of Bifidobacterium Bifidum PRL2010 Reveals Metabolic Pathways for Host-Derived Glycan Foraging. Proc. Natl. Acad. Sci. USA 2010, 107, 19514–19519. [Google Scholar] [CrossRef] [Green Version]
- Sela, D.A.; Chapman, J.; Adeuya, A.; Kim, J.H.; Chen, F.; Whitehead, T.R.; Lapidus, A.; Rokhsar, D.S.; Lebrilla, C.B.; German, J.B.; et al. The Genome Sequence of Bifidobacterium Longum Subsp. Infantis Reveals Adaptations for Milk Utilization within the Infant Microbiome. Proc. Natl. Acad. Sci. USA 2008, 105, 18964–18969. [Google Scholar] [CrossRef] [Green Version]
- Arboleya, S.; Bottacini, F.; O’Connell-Motherway, M.; Ryan, C.A.; Ross, R.P.; van Sinderen, D.; Stanton, C. Gene-Trait Matching across the Bifidobacterium Longum Pan-Genome Reveals Considerable Diversity in Carbohydrate Catabolism among Human Infant Strains. BMC Genom. 2018, 19, 33. [Google Scholar] [CrossRef]
- Duranti, S.; Milani, C.; Lugli, G.A.; Mancabelli, L.; Turroni, F.; Ferrario, C.; Mangifesta, M.; Viappiani, A.; Sánchez, B.; Margolles, A.; et al. Evaluation of Genetic Diversity among Strains of the Human Gut Commensal Bifidobacterium Adolescentis. Sci. Rep. 2016, 6, 23971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, W.; Pei, Z.; Zang, M.; Lee, Y.-K.; Zhao, J.; Chen, W.; Wang, H.; Zhang, H. Comparative Genomic Analysis of Bifidobacterium Bifidum Strains Isolated from Different Niches. Genes 2021, 12, 1504. [Google Scholar] [CrossRef]
- McGuire, M.K.; Meehan, C.L.; McGuire, M.A.; Williams, J.E.; Foster, J.; Sellen, D.W.; Kamau-Mbuthia, E.W.; Kamundia, E.W.; Mbugua, S.; Moore, S.E.; et al. What’s Normal? Oligosaccharide Concentrations and Profiles in Milk Produced by Healthy Women Vary Geographically. Am. J. Clin. Nutr. 2017, 105, 1086–1100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gotoh, A.; Katoh, T.; Sugiyama, Y.; Kurihara, S.; Honda, Y.; Sakurama, H.; Kambe, T.; Ashida, H.; Kitaoka, M.; Yamamoto, K.; et al. Novel Substrate Specificities of Two Lacto-N-Biosidases towards β-Linked Galacto-N-Biose-Containing Oligosaccharides of Globo H, Gb5, and GA1. Carbohydr. Res. 2015, 408, 18–24. [Google Scholar] [CrossRef]
- Spichtig, V.; Michaud, J.; Austin, S. Determination of Sialic Acids in Milks and Milk-Based Products. Anal. Biochem. 2010, 405, 28–40. [Google Scholar] [CrossRef] [PubMed]
- Kiyohara, M.; Tanigawa, K.; Chaiwangsri, T.; Katayama, T.; Ashida, H.; Yamamoto, K. An Exo-Alpha-Sialidase from Bifidobacteria Involved in the Degradation of Sialyloligosaccharides in Human Milk and Intestinal Glycoconjugates. Glycobiology 2011, 21, 437–447. [Google Scholar] [CrossRef] [Green Version]
- Mao, B.; Li, D.; Ai, C.; Zhao, J.; Zhang, H.; Chen, W. Lactulose Differently Modulates the Composition of Luminal and Mucosal Microbiota in C57BL/6J Mice. J. Agric. Food Chem. 2016, 64, 6240–6247. [Google Scholar] [CrossRef] [PubMed]
- Luo, R.; Liu, B.; Xie, Y.; Li, Z.; Huang, W.; Yuan, J.; He, G.; Chen, Y.; Pan, Q.; Liu, Y.; et al. SOAPdenovo2: An Empirically Improved Memory-Efficient Short-Read de Novo Assembler. GigaScience 2012, 1, 18. [Google Scholar] [CrossRef]
- Pan, Q.; Cen, S.; Yu, L.; Tian, F.; Zhao, J.; Zhang, H.; Chen, W.; Zhai, Q. Niche-Specific Adaptive Evolution of Lactobacillus Plantarum Strains Isolated from Human Feces and Paocai. Front. Cell. Infect. Microbiol. 2020, 10, 615876. [Google Scholar] [CrossRef] [PubMed]
- Borodovsky, M.; Mills, R.; Besemer, J.; Lomsadze, A. Prokaryotic Gene Prediction Using GeneMark and GeneMark.Hmm. Curr. Protoc. Bioinform. 2003, 1, 4–5. [Google Scholar] [CrossRef]
- Fischer, S.; Brunk, B.P.; Chen, F.; Gao, X.; Harb, O.S.; Iodice, J.B.; Shanmugam, D.; Roos, D.S.; Stoeckert, C.J., Jr. Using OrthoMCL to Assign Proteins to OrthoMCL-DB Groups or to Cluster Proteomes into New Ortholog Groups. Curr. Protoc. Bioinform. 2011, 1, 4.5.1–4.5.16. [Google Scholar] [CrossRef] [Green Version]
- Cantarel, B.L.; Coutinho, P.M.; Rancurel, C.; Bernard, T.; Lombard, V.; Henrissat, B. The Carbohydrate-Active EnZymes Database (CAZy): An Expert Resource for Glycogenomics. Nucleic Acids Res. 2009, 37, D233–D238. [Google Scholar] [CrossRef]
- Jia, B.; Raphenya, A.R.; Alcock, B.; Waglechner, N.; Guo, P.; Tsang, K.K.; Lago, B.A.; Dave, B.M.; Pereira, S.; Sharma, A.N.; et al. CARD 2017: Expansion and Model-Centric Curation of the Comprehensive Antibiotic Resistance Database. Nucleic Acids Res. 2017, 45, D566–D573. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.; Luo, B.; Cai, J.; Yang, B.; Zhang, Y.; Tian, F.; Ni, Y. Evaluation of Indigenous Lactic Acid Bacteria of Raw Mare Milk from Pastoral Areas in Xinjiang, China, for Potential Use in Probiotic Fermented Dairy Products. J. Dairy Sci. 2021, 104, 5166–5184. [Google Scholar] [CrossRef]
- Deng, W.; Wang, Y.; Liu, Z.; Cheng, H.; Xue, Y. HemI: A Toolkit for Illustrating Heatmaps. PLoS ONE 2014, 9, e111988. [Google Scholar] [CrossRef] [PubMed]
- Sivan, A.; Corrales, L.; Hubert, N.; Williams, J.B.; Aquino-Michaels, K.; Earley, Z.M.; Benyamin, F.W.; Lei, Y.M.; Jabri, B.; Alegre, M.-L.; et al. Commensal Bifidobacterium Promotes Antitumor Immunity and Facilitates Anti-PD-L1 Efficacy. Science 2015, 350, 1084–1089. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reiff, C.; Kelly, D. Inflammatory Bowel Disease, Gut Bacteria and Probiotic Therapy. Int. J. Med. Microbiol. 2010, 300, 25–33. [Google Scholar] [CrossRef]
- Singh, J.; Rivenson, A.; Tomita, M.; Shimamura, S.; Ishibashi, N.; Reddy, B.S. Bifidobacterium Longum, a Lactic Acid-Producing Intestinal Bacterium Inhibits Colon Cancer and Modulates the Intermediate Biomarkers of Colon Carcinogenesis. Carcinogenesis 1997, 18, 833–841. [Google Scholar] [CrossRef]
- Hidalgo-Cantabrana, C.; Delgado, S.; Ruiz, L.; Ruas-Madiedo, P.; Sánchez, B.; Margolles, A. Bifidobacteria and Their Health-Promoting Effects. Microbiol. Spectr. 2017, 5, 73–89. [Google Scholar] [CrossRef]
- Milani, C.; Mancabelli, L.; Lugli, G.A.; Duranti, S.; Turroni, F.; Ferrario, C.; Mangifesta, M.; Viappiani, A.; Ferretti, P.; Gorfer, V.; et al. Exploring Vertical Transmission of Bifidobacteria from Mother to Child. Appl. Environ. Microbiol. 2015, 81, 7078–7087. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Milani, C.; Duranti, S.; Bottacini, F.; Casey, E.; Turroni, F.; Mahony, J.; Belzer, C.; Delgado Palacio, S.; Arboleya Montes, S.; Mancabelli, L.; et al. The First Microbial Colonizers of the Human Gut: Composition, Activities, and Health Implications of the Infant Gut Microbiota. Microbiol. Mol. Biol. Rev. 2017, 81, e00036-17. [Google Scholar] [CrossRef] [Green Version]
- Ishikawa, E.; Matsuki, T.; Kubota, H.; Makino, H.; Sakai, T.; Oishi, K.; Kushiro, A.; Fujimoto, J.; Watanabe, K.; Watanuki, M.; et al. Ethnic Diversity of Gut Microbiota: Species Characterization of Bacteroides Fragilis Group and Genus Bifidobacterium in Healthy Belgian Adults, and Comparison with Data from Japanese Subjects. J. Biosci. Bioeng. 2013, 116, 265–270. [Google Scholar] [CrossRef] [PubMed]
- Schroeder, B.O.; Birchenough, G.M.H.; Ståhlman, M.; Arike, L.; Johansson, M.E.V.; Hansson, G.C.; Bäckhed, F. Bifidobacteria or Fiber Protects against Diet-Induced Microbiota-Mediated Colonic Mucus Deterioration. Cell Host Microbe 2018, 23, 27–40.e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arboleya, S.; Watkins, C.; Stanton, C.; Ross, R.P. Gut Bifidobacteria Populations in Human Health and Aging. Front. Microbiol. 2016, 7, 1204. [Google Scholar] [CrossRef] [Green Version]
- Ward, R.E.; Ninonuevo, M.; Mills, D.A.; Lebrilla, C.B.; German, J.B. In Vitro Fermentability of Human Milk Oligosaccharides by Several Strains of Bifidobacteria. Mol. Nutr. Food Res. 2007, 51, 1398–1405. [Google Scholar] [CrossRef]
- Garron, M.-L.; Henrissat, B. The Continuing Expansion of CAZymes and Their Families. Curr. Opin. Chem. Biol. 2019, 53, 82–87. [Google Scholar] [CrossRef]
- He, B.; Jin, S.; Cao, J.; Mi, L.; Wang, J. Metatranscriptomics of the Hu Sheep Rumen Microbiome Reveals Novel Cellulases. Biotechnol. Biofuels 2019, 12, 153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Den Broek, L.A.M.; Voragen, A.G.J. Bifidobacterium Glycoside Hydrolases and (Potential) Prebiotics. Innov. Food Sci. Emerg. Technol. 2008, 9, 401–407. [Google Scholar] [CrossRef]
- Egan, M.; Motherway, M.O.; Ventura, M.; van Sinderen, D. Metabolism of Sialic Acid by Bifidobacterium Breve UCC2003. Appl. Environ. Microbiol. 2014, 80, 4414–4426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ashida, H.; Miyake, A.; Kiyohara, M.; Wada, J.; Yoshida, E.; Kumagai, H.; Katayama, T.; Yamamoto, K. Two Distinct Alpha-L-Fucosidases from Bifidobacterium Bifidum Are Essential for the Utilization of Fucosylated Milk Oligosaccharides and Glycoconjugates. Glycobiology 2009, 19, 1010–1017. [Google Scholar] [CrossRef] [Green Version]
- Yu, Z.-T.; Chen, C.; Newburg, D.S. Utilization of Major Fucosylated and Sialylated Human Milk Oligosaccharides by Isolated Human Gut Microbes. Glycobiology 2013, 23, 1281–1292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bondue, P.; Crèvecoeur, S.; Brose, F.; Daube, G.; Seghaye, M.-C.; Griffiths, M.W.; LaPointe, G.; Delcenserie, V. Cell-Free Spent Media Obtained from Bifidobacterium Bifidum and Bifidobacterium Crudilactis Grown in Media Supplemented with 3′-Sialyllactose Modulate Virulence Gene Expression in Escherichia Coli O157:H7 and Salmonella Typhimurium. Front. Microbiol. 2016, 7, 1460. [Google Scholar] [CrossRef] [Green Version]
- Duranti, S.; Milani, C.; Lugli, G.A.; Turroni, F.; Mancabelli, L.; Sanchez, B.; Ferrario, C.; Viappiani, A.; Mangifesta, M.; Mancino, W.; et al. Insights from Genomes of Representatives of the Human Gut Commensal Bifidobacterium Bifidum. Environ. Microbiol. 2015, 17, 2515–2531. [Google Scholar] [CrossRef]
- Chun, J.; Oren, A.; Ventosa, A.; Christensen, H.; Arahal, D.R.; da Costa, M.S.; Rooney, A.P.; Yi, H.; Xu, X.-W.; De Meyer, S.; et al. Proposed Minimal Standards for the Use of Genome Data for the Taxonomy of Prokaryotes. Int. J. Syst. Evol. Microbiol. 2018, 68, 461–466. [Google Scholar] [CrossRef] [PubMed]
- Ciufo, S.; Kannan, S.; Sharma, S.; Badretdin, A.; Clark, K.; Turner, S.; Brover, S.; Schoch, C.L.; Kimchi, A.; DiCuccio, M. Using Average Nucleotide Identity to Improve Taxonomic Assignments in Prokaryotic Genomes at the NCBI. Int. J. Syst. Evol. Microbiol. 2018, 68, 2386–2392. [Google Scholar] [CrossRef] [PubMed]
- Sela, D.A. Bifidobacterial Utilization of Human Milk Oligosaccharides. Int. J. Food Microbiol. 2011, 149, 58–64. [Google Scholar] [CrossRef] [PubMed]
- Turroni, F.; Milani, C.; van Sinderen, D.; Ventura, M. Genetic Strategies for Mucin Metabolism in Bifidobacterium Bifidum PRL2010: An Example of Possible Human-Microbe Co-Evolution. Gut Microbes 2011, 2, 183–189. [Google Scholar] [CrossRef] [PubMed] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Demographic Data | Age Group (y) | Values or No. (%) | ||||
|---|---|---|---|---|---|---|
| 0–17 | 18–65 | 66–108 | 0–17 | 18–65 | 66–108 | |
| Gender | ||||||
| Male | 96 | 80 | 33 | 96 (51%) | 80 (39%) | 33 (35%) |
| Female | 69 | 118 | 61 | 69 (37%) | 118 (58%) | 61 (65%) |
| Not specified | 23 | 6 | 0 | 23 (12%) | 6 (3%) | 0 |
| Total no. of samples | 188 | 204 | 94 | – | ||
| Total no. of strains | 260 | 244 | 106 | – | ||
| Strain | Age 0−17 | Age 18−65 | Age 66−108 | |||
|---|---|---|---|---|---|---|
| B. bifidum | FJSNJ1M3 | ++ | FGSZY50M8 | – | FHNFQ34M6 | + |
| FZJHZD4M4 | +++ | FHNFQ25M12 | − | FHNFQ11M4 | ++ | |
| FAHWH21M3 | ++ | FGSYC45M3 | − | FHNXY17M1 | + | |
| B. longum subsp. infantis | FGZ19I1M3 | +++ | FGZ6I2M6 | +++ | / | |
| HeNJZ8M1 | +++ | FJ12WI1M14 | +++ | / | ||
| FGZ17I1M1 | +++ | FJ12WI1M1 | ++ | / | ||
| B. breve | JSWX17M1 | ++ | / | FHNXY48M6 | +++ | |
| FJSZJ1M5 | ++ | / | FCQNA20M1 | ++ | ||
| FBJCP1M6 | + | / | FHNFQ34M6 | ++ | ||
| Host | Bacterial Strain | Gene No. | Genome Size/Mb | G + C% | Level | Accession Number |
|---|---|---|---|---|---|---|
| Human feces (0–17) | B. bifidum FJSNJ1M3 | 1910 | 2.13 | 62.7 | Scaffold | SRR13205608 |
| B. bifidum FZJHZD4M4 | 1959 | 2.17 | 62.6 | Scaffold | SRR13205566 | |
| B. bifidum FAHWH21M3 | 1899 | 2.12 | 62.7 | Scaffold | SRR13205659 | |
| Human feces (18–65) | B. bifidum FGSZY50M8 | 1956 | 2.18 | 62.5 | Scaffold | SRR13205646 |
| B. bifidum FHNFQ25M12 | 1985 | 2.12 | 62.6 | Scaffold | SRR13205635 | |
| B. bifidum FGSYC45M3 | 1932 | 2.13 | 62.7 | Scaffold | SRR13205647 |
| Strains | PEN | AMS | AMO | CLM | RIF | CEF | TET | TEC | VAN | OXA | CIP | GM | T/S | NEO | KAN | S | ATM | E |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| FJSNJ1M3 | S | S | S | S | S | S | S | S | S | S | R | S | R | R | R | R | R | R |
| FZJHZD4M4 | S | S | S | S | S | S | S | S | S | S | R | R | R | R | R | R | R | R |
| FAHWH21M3 | S | S | S | S | S | S | S | S | S | S | R | R | R | R | R | R | R | R |
| FHNFQ25M12 | S | S | S | S | S | S | S | S | S | S | R | R | R | R | R | R | R | R |
| FGSZY50M8 | S | S | S | S | S | S | S | S | S | S | R | S | R | R | R | R | R | R |
| FGSYC45M3 | S | S | S | S | S | S | S | S | S | S | R | R | R | R | R | R | R | R |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wei, X.; Yu, L.; Zhang, C.; Ni, Y.; Zhang, H.; Zhai, Q.; Tian, F. Genetic-Phenotype Analysis of Bifidobacterium bifidum and Its Glycoside Hydrolase Gene Distribution at Different Age Groups. Foods 2023, 12, 922. https://doi.org/10.3390/foods12050922
Wei X, Yu L, Zhang C, Ni Y, Zhang H, Zhai Q, Tian F. Genetic-Phenotype Analysis of Bifidobacterium bifidum and Its Glycoside Hydrolase Gene Distribution at Different Age Groups. Foods. 2023; 12(5):922. https://doi.org/10.3390/foods12050922
Chicago/Turabian StyleWei, Xiaojing, Leilei Yu, Chuan Zhang, Yongqing Ni, Hao Zhang, Qixiao Zhai, and Fengwei Tian. 2023. "Genetic-Phenotype Analysis of Bifidobacterium bifidum and Its Glycoside Hydrolase Gene Distribution at Different Age Groups" Foods 12, no. 5: 922. https://doi.org/10.3390/foods12050922