

Simultaneous Determination of Neonicotinoid and Carbamate Pesticides in Freeze-Dried Cabbage by Modified QuEChERS and Ultra-Performance Liquid Chromatography–Tandem Mass Spectrometry


 ,
, 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Reagents and Materials
2.2. Pesticide-Free FD Cabbage Preparation
2.3. Sample Fortification
2.4. Real Sample Analysis
2.5. Instrument and Apparatus
2.6. Method Validation
2.7. Post-Column Infusion
3. Results and Discussion
3.1. Optimization of the QuEChERS Method
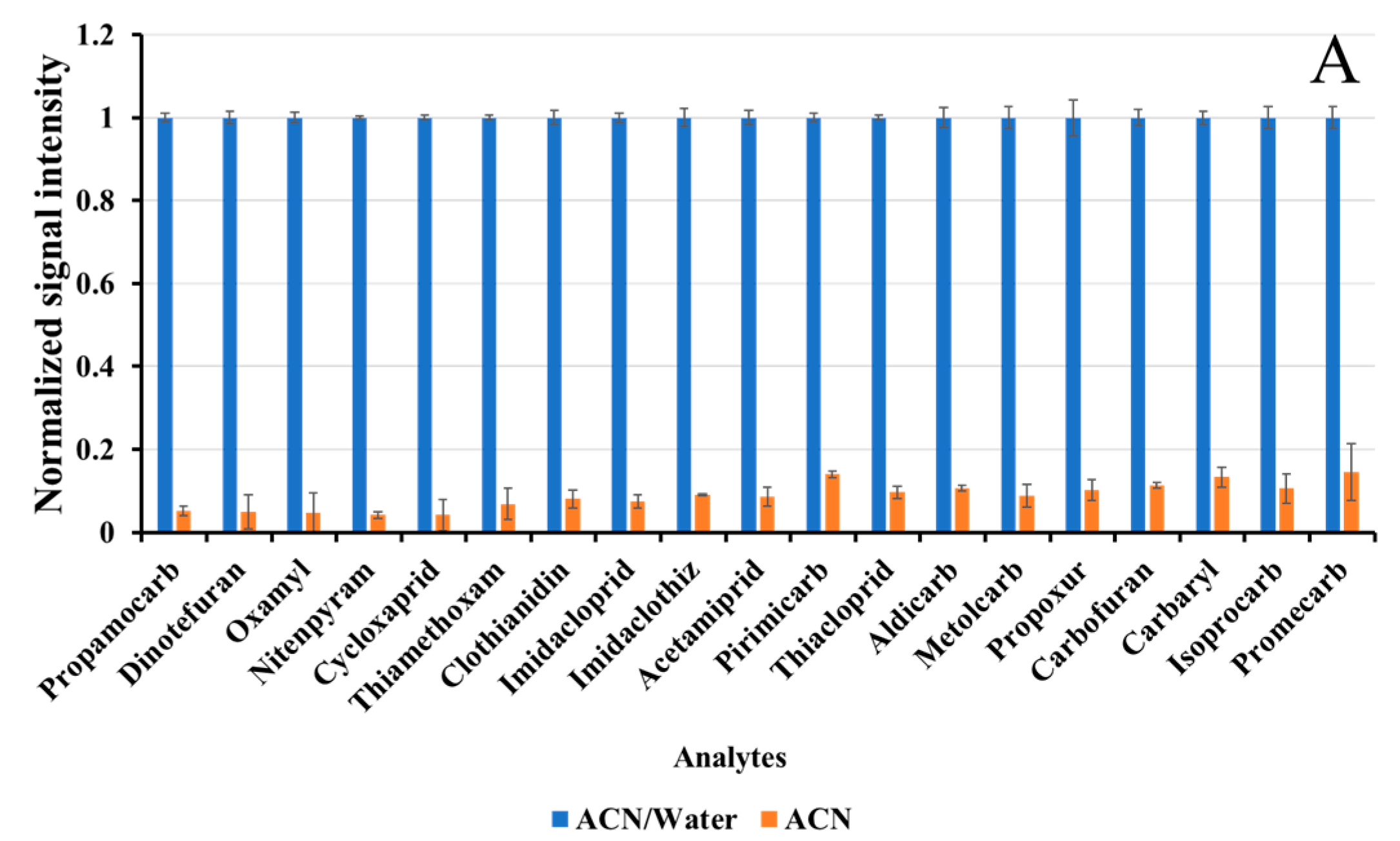
3.1.1. Sample Extraction
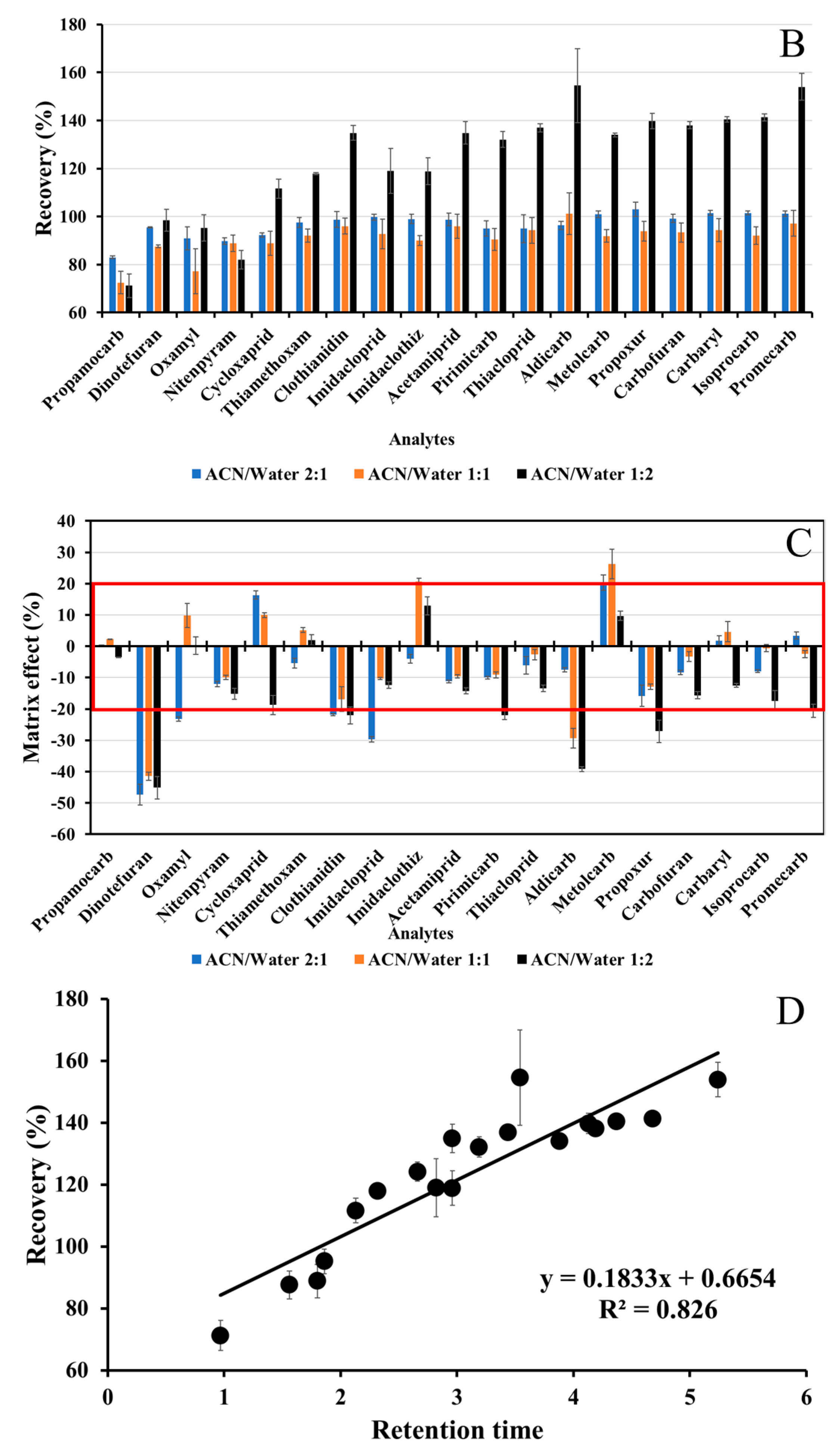
3.1.2. Liquid–Liquid Phase Partitioning
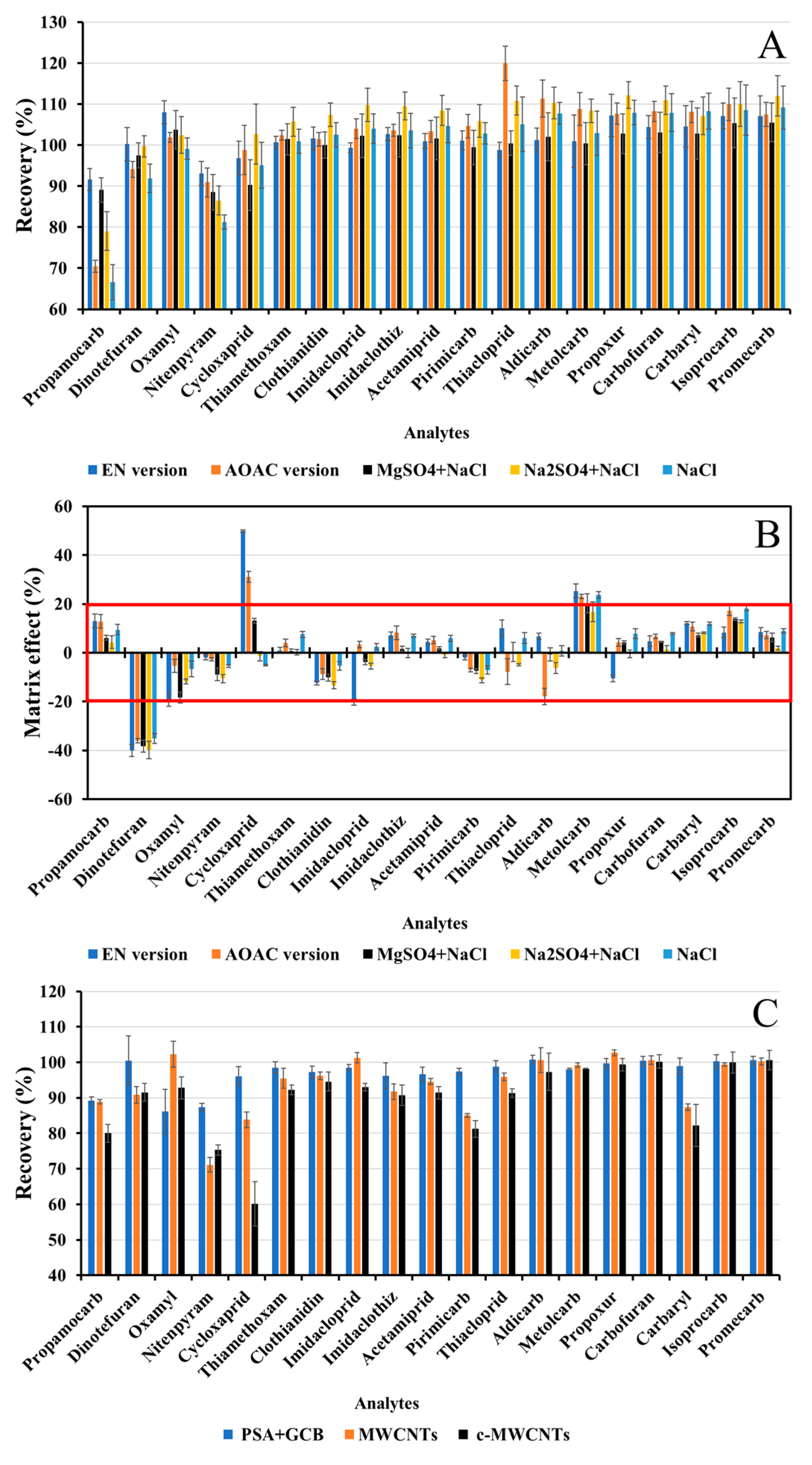
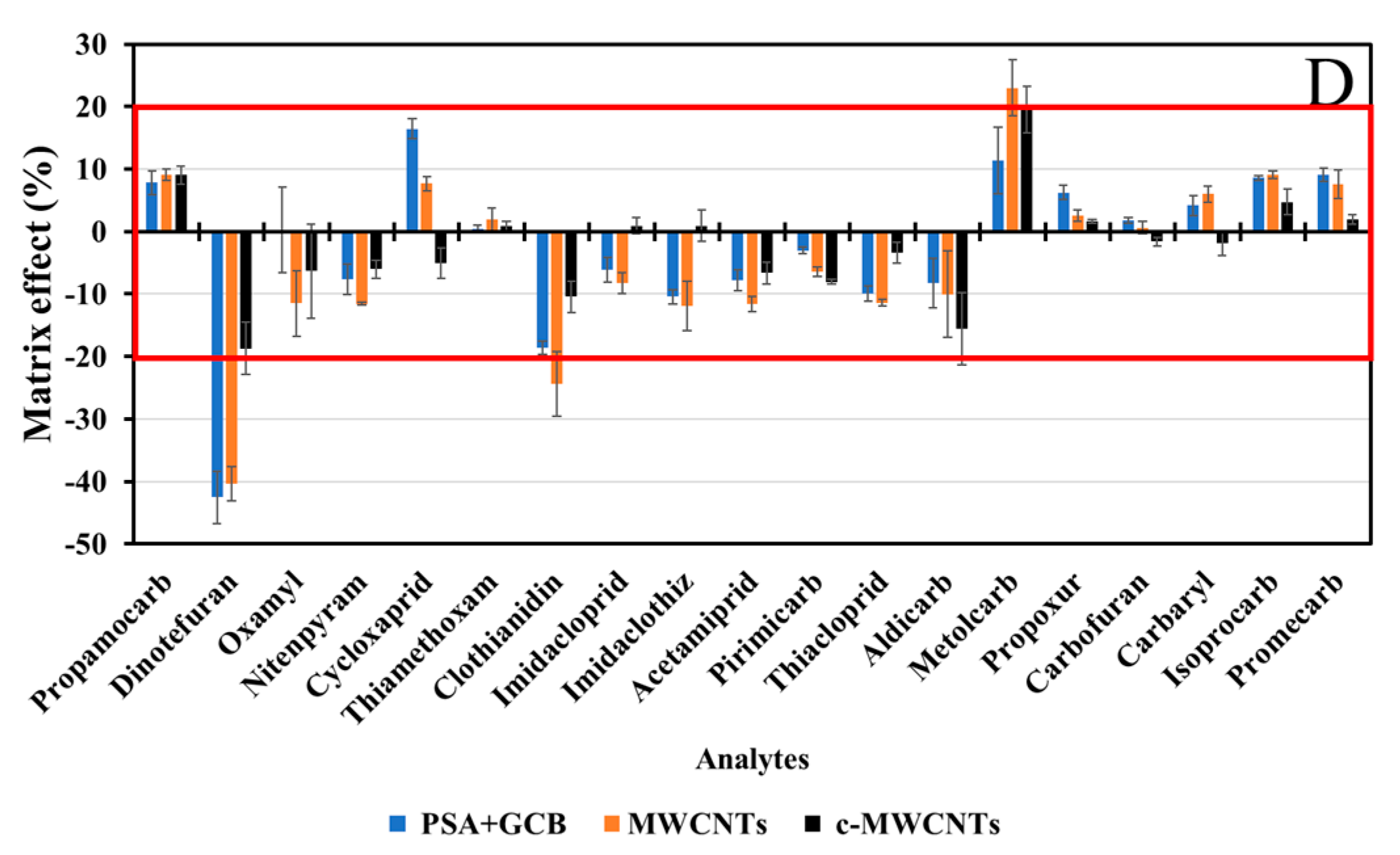
3.1.3. Sample Clean-Up
3.2. Optimization of UPLC–MS/MS/MS Conditions
3.3. Method Validation
3.3.1. LODs, LOQs, and Linearity
3.3.2.Assay Selectivity
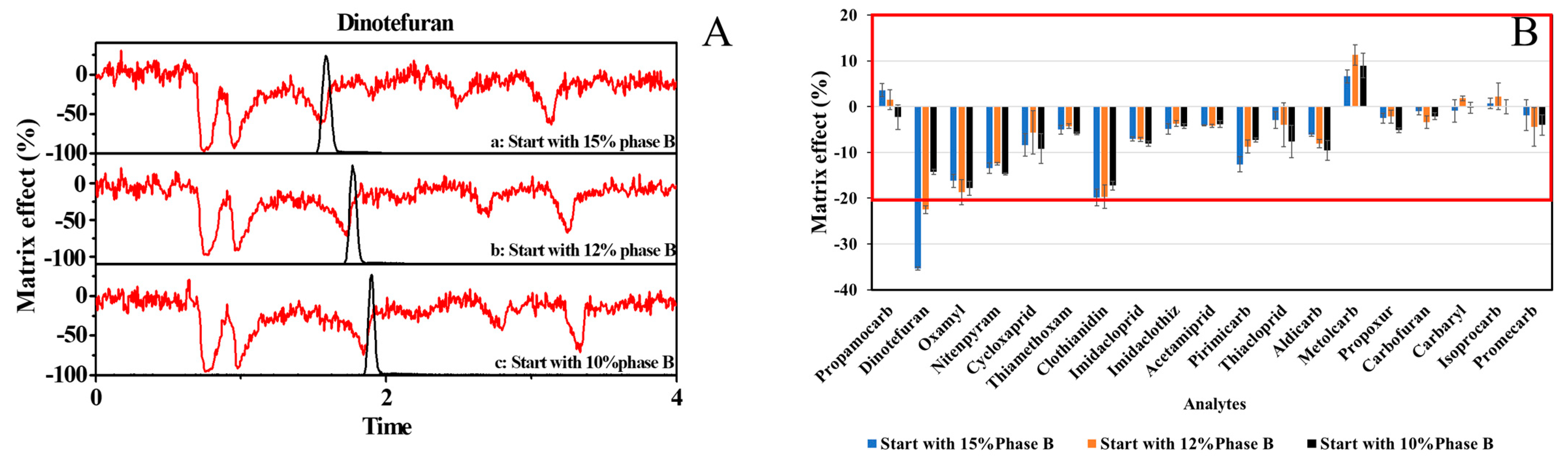
3.3.3. Matrix Effect
3.3.4. Recovery and Repeatability
3.3.5. Stability of Final Extracts
3.4. Real Sample Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Jin, X.; Oliviero, T.; van der Sman, R.G.M.; Verkerk, R.; Dekker, M.; van Boxtel, A.J.B. Impact of different drying trajectories on degradation of nutritional compounds in broccoli (Brassica oleracea var. italica). LWT Food Sci. Technol. 2014, 59, 189–195. [Google Scholar] [CrossRef]
- Rajkumar, G.; Shanmugam, S.; Galvâo, M.D.S.; Dutra Sandes, R.D.; Leite Neta, M.T.S.; Narain, N.; Mujumdar, A.S. Comparative evaluation of physical properties and volatiles profile of cabbages subjected to hot air and freeze drying. LWT Food Sci. Technol. 2017, 80, 501–509. [Google Scholar] [CrossRef]
- Xu, Y.; Xiao, Y.; Lagnika, C.; Li, D.; Liu, C.; Jiang, N.; Song, J.; Zhang, M. A comparative evaluation of nutritional properties, antioxidant capacity and physical characteristics of cabbage (Brassica oleracea var. Capitate var L.) subjected to different drying methods. Food Chem. 2020, 309, 124935. [Google Scholar] [CrossRef]
- NASA Technology Transfer Program. Freeze-Dried Foods Nourish Adventurers and the Imagination. Available online: https://spinoff.nasa.gov/Spinoff2020/cg_2.html, 2020 (accessed on 1 October 2022).
- Bhatta, S.; Stevanovic Janezic, T.; Ratti, C. Freeze-Drying of Plant-Based Foods. Foods 2020, 9, 87. [Google Scholar] [CrossRef]
- Kim, J.-H.; Choi, S.-G.; Kwon, Y.S.; Hong, S.-M.; Seo, J.-S. Development of cabbage reference material for multi-residue pesticide analysis. Appl. Biol. Chem. 2017, 61, 15–23. [Google Scholar] [CrossRef]
- Yang, B.; Ma, W.; Wang, S.; Shi, L.; Li, X.; Ma, Z.; Zhang, Q.; Li, H. Determination of eight neonicotinoid insecticides in Chinese cabbage using a modified QuEChERS method combined with ultra performance liquid chromatography-tandem mass spectrometry. Food Chem. 2022, 387, 132935. [Google Scholar] [CrossRef]
- Kecojević, I.; Đekić, S.; Lazović, M.; Mrkajić, D.; Baošić, R.; Lolić, A. Evaluation of LC-MS/MS methodology for determination of 179 multi-class pesticides in cabbage and rice by modified QuEChERS extraction. Food Control 2021, 123, 107693. [Google Scholar] [CrossRef]
- Cesarino, I.; Moraes, F.C.; Lanza, M.R.V.; Machado, S.A.S. Electrochemical detection of carbamate pesticides in fruit and vegetables with a biosensor based on acetylcholinesterase immobilised on a composite of polyaniline–carbon nanotubes. Food Chem. 2012, 135, 873–879. [Google Scholar] [CrossRef] [PubMed]
- Cimino, A.M.; Boyles, A.L.; Thayer, K.A.; Perry, M.J. Effects of Neonicotinoid Pesticide Exposure on Human Health: A Systematic Review. Environ. Health Persp. 2017, 125, 155–162. [Google Scholar] [CrossRef] [PubMed]
- Marfo, J.T.; Fujioka, K.; Ikenaka, Y.; Nakayama, S.M.M.; Mizukawa, H.; Aoyama, Y.; Ishizuka, M.; Taira, K. Relationship between Urinary N-Desmethyl-Acetamiprid and Typical Symptoms including Neurological Findings: A Prevalence Case-Control Study. PLoS ONE 2015, 10, e0142172. [Google Scholar] [CrossRef] [Green Version]
- Li, Q.; Kobayashi, M.; Kawada, T. Effects of carbamate pesticides on human natural killers and T lymphocytes. Toxicol. Lett. 2015, 238, S217–S218. [Google Scholar] [CrossRef]
- European Food Safety Authority. Pesticide Residues and the MRLs That Apply for Such Residues in Food Products. 2022. Available online: http://ec.europa.eu/food/plant/pesticides/eu-pesticides-database/public/?event=homepage&language=EN (accessed on 1 October 2022).
- NHC, MoA & SAMR of China. Maximum Residue Limits for Pesticides in Food. 2021. Available online: https://www.sdtdata.com/fx/fcv1/ts LibCard/183688.html (accessed on 3 March 2021).
- The Electronic Code of Federal Regulations. Tolerances and Exemptions for Pesticide Chemical Residues in Food. 2022. Available online: https://www.ecfr.gov/cgi-bin/text-idx?c=ecfr&sid=1a0ecaf51aa3dba662c9cbf1b4336eaf&tpl=/ecfrbrowse/Title40/40cfr180_main_02.tpl (accessed on 1 October 2022).
- Ma, L.; Wang, Y.; Li, H.; Peng, F.; Qiu, B.; Yang, Z. Development of QuEChERS-DLLME method for determination of neonicotinoid pesticide residues in grains by liquid chromatography-tandem mass spectrometry. Food Chem. 2020, 331, 127190. [Google Scholar] [CrossRef] [PubMed]
- Végh, R.; Sörös, C.; Majercsik, N.; Sipos, L. Determination of Pesticides in Bee Pollen: Validation of a Multiresidue High-Performance Liquid Chromatography-Mass Spectrometry/Mass Spectrometry Method and Testing Pollen Samples of Selected Botanical Origin. J. Agric. Food Chem. 2022, 70, 1507–1515. [Google Scholar] [CrossRef]
- Guan, K.; Huang, R.; Liu, H.; Huang, Y.; Chen, A.; Zhao, X.; Wang, S.; Zhang, L. Development of a Reliable ic-ELISA with a Robust Antimatrix Interference Capability Based on QuEChERS Technology for the Rapid Detection of Zearalenone in Edible and Medical Coix Seeds and Subsequent Risk Assessments. Foods 2022, 11, 2983. [Google Scholar] [CrossRef]
- Octanol/Water Partition Coefficient. Available online: http://www.chemspider.com/ (accessed on 1 October 2022).
- Dos Santos, E.O.; Gonzales, J.O.; Ores, J.C.; Marube, L.C.; Caldas, S.S.; Furlong, E.B.; Primel, E.G. Sand as a solid support in ultrasound-assisted MSPD: A simple, green and low-cost method for multiresidue pesticide determination in fruits and vegetables. Food Chem. 2019, 297, 124926. [Google Scholar] [CrossRef] [PubMed]
- Farouk, M.; Hussein, L.A.E.A.; El Azab, N.F. Simultaneous determination of three neonicotinoid insecticide residues and their metabolite in cucumbers and soil by QuEChERS clean up and liquid chromatography with diode-array detection. Anal. Methods 2016, 8, 4563–4575. [Google Scholar] [CrossRef]
- Ma, W.; Yang, B.; Li, J.; Li, X. Amino-functional metal-organic framework as a general applicable adsorbent for simultaneous enrichment of nine neonicotinoids. Chem. Eng. J. 2022, 434, 134629. [Google Scholar] [CrossRef]
- Zuo, J.; Cai, R.; An, Y.; Tang, H. Simultaneous Quantification of Five Stereoisomeric Hexoses in Nine Biological Matrices Using Ultrahigh Performance Liquid Chromatography with Tandem Mass Spectrometry. J. Anal. Test. 2020, 4, 249–256. [Google Scholar] [CrossRef]
- Li, X.; Ma, W.; Zhang, Q.; Li, H.; Liu, H. Determination of patulin in apple juice by amine-functionalized solid-phase extraction coupled with isotope dilution liquid chromatography tandem mass spectrometry. J. Sci. Food Agric. 2021, 101, 1767–1771. [Google Scholar] [CrossRef]
- Ma, W.; Li, J.; Li, X.; Liu, H. Enrichment of diamide insecticides from environmental water samples using metal-organic frameworks as adsorbents for determination by liquid chromatography tandem mass spectrometry. J. Hazard. Mater. 2022, 422, 126839. [Google Scholar] [CrossRef]
- European Commission. Guidance Document on Analytical Quality Control and Method Validation for Pesticide Residues Analysis in Food and Feed SANTE 11312/2021. 2021. Available online: https://ec.europa.eu/food/system/files/2022-02/pesticides_mrl_guidelines_wrkdoc_2021-11312.pdf (accessed on 24 February 2021).
- Stahnke, H.; Reemtsma, T.; Alder, L. Cocompensation of Matrix Effects by Postcolumn Infusion of a Monitor Substance in Multiresidue Analysis with LC-MS/MS. Anal. Chem. 2009, 81, 2185–2192. [Google Scholar] [CrossRef] [PubMed]
- Lehotay, S.J.; Sapozhnikova, Y.; Han, L.; Johnston, J.J. Analysis of Nitrosamines in Cooked Bacon by QuEChERS Sample Preparation and Gas Chromatography-Tandem Mass Spectrometry with Backflushing. J. Agric. Food Chem. 2015, 63, 10341–10351. [Google Scholar] [CrossRef]
- Mastovska, K.; Dorweiler, K.J.; Lehotay, S.J.; Wegscheid, J.S.; Szpylka, K.A. Pesticide multiresidue analysis in cereal grains using modified QuEChERS method combined with automated direct sample introduction GC-TOFMS and UPLC-MS/MS techniques. J. Agric. Food Chem. 2010, 58, 5959–5972. [Google Scholar] [CrossRef]
- Zhao, W.; Shi, Y. A porous boron nitride nanorods-based QuEChERS analysis method for detection of five neonicotinoid pesticide residues in goji berries. J. Chromatogr. A 2022, 1670, 462968. [Google Scholar] [CrossRef] [PubMed]
- Anastassiades, M.; Lehotay, S.J.; Štajnbaher, D.; Schenck, F.J. Fast and Easy multiresidue method employing acetonitrile extraction partitioning and “dispersive solid-phase extraction” for the determination of pesticide residues in produce. J. AOAC Int. 2003, 86, 412–431. [Google Scholar] [CrossRef] [PubMed]
- Islam, A.K.M.M.; Noh, H.H.; Ro, J.-H.; Kim, D.; Oh, M.-S.; Son, K.; Kwon, H. Optimization and validation of a method for the determination of acidic pesticides in cabbage and spinach by modifying QuEChERS procedure and liquid chromatography-tandem mass spectrometry. J. Chromatogr. B 2021, 1173, 122667. [Google Scholar] [CrossRef]
- Wang, S.; Li, M.; Li, X.; Li, X.; Li, X.; Li, S.; Zhang, Q.; Li, H. A functionalized carbon nanotube nanohybrids-based QuEChERS method for detection of pesticide residues in vegetables and fruits. J. Chromatogr. A 2020, 1631, 461526. [Google Scholar] [CrossRef]
- Thurman, E.M.; Ferrer, I.; Barceló, D. Choosing between Atmospheric Pressure Chemical Ionization and Electrospray Ionization Interfaces for the HPLC/MS Analysis of Pesticides. Anal. Chem. 2001, 73, 5441–5449. [Google Scholar] [CrossRef]
- Ikonomou, M.G.; Blades, A.T.; Kebarle, P. Investigations of the Electrospray Interface for Liquid Chromatography/Mass Spectrometry. Anal. Chem. 1990, 62, 957–967. [Google Scholar] [CrossRef]
- Yang, X.J.; Qu, Y.; Yuan, Q.; Wan, P.; Du, Z.; Chen, D.; Wong, C. Effect of ammonium on liquid- and gas-phase protonation and deprotonation in electrospray ionization mass spectrometry. Analyst 2013, 138, 659–665. [Google Scholar] [CrossRef]
- Ministry of Agriculture and Rural Affairs of People’s Repbulic of China. List of Prohibited Pesticides. Available online: http://www.zzys.moa.gov.cn/gzdt/201911/t20191129_6332604.htm (accessed on 19 January 2023).
- Kunpatee, K.; Kalcher, K.; Chailapakul, O.; Chaiyo, S.; Samphao, A. A paper chromatographic- based electrochemical analytical device for the separation and simultaneous detection of carbofuran and carbaryl pesticides. Sensor. Actuat. B Chem. 2023, 377, 133116. [Google Scholar] [CrossRef]
- Wang, M.; Wang, J.; Wang, K.; Zhang, L.; Cao, X.; Guo, C.; Wang, J.; Wu, B. Magnetic mesoporous material derived from MIL-88B modified by l-alanine as modified QuEChERS adsorbent for the determination of 6 pesticide residues in 4 vegetables by UPLC-MS/MS. Food Chem. 2022, 384, 132325. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Xu, J.; Dong, F.; Liu, X.; Li, X.; Li, Y.; Wu, X.; Liang, X.; Zheng, Y. Simultaneous determination of four neonicotinoid insecticides residues in cereals, vegetables and fruits using ultra-performance liquid chromatography/tandem mass spectrometry. Anal. Methods 2013, 5, 1449–1455. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Analytes | Rt (a) (min) | CV (b) (V) | Precursor Ion (m/z) | Product Ion 1 (m/z) | CE (c)-1 (V) | Product Ion 2 (m/z) | CE-2 (V) |
|---|---|---|---|---|---|---|---|
| Propamocarb | 1.71 | 35 | 189.0 | 102.0 | 17 | 144.0 | 10 |
| Dinotefuran | 1.85 | 30 | 203.0 | 129.0 | 10 | 113.3 | 10 |
| Oxamyl | 2.15 | 65 | 242.0 | 72.0 | 10 | 121.0 | 10 |
| Nitenpyram | 2.18 | 30 | 271.0 | 56.1 | 24 | 126.0 | 32 |
| Cycloxaprid | 2.56 | 30 | 323.0 | 125.8 | 36 | 150.9 | 22 |
| Thiamethoxam | 2.58 | 25 | 291.9 | 210.9 | 12 | 132.0 | 22 |
| Clothianidin | 2.89 | 25 | 250.1 | 168.9 | 12 | 132.0 | 14 |
| Imidacloprid | 3.04 | 35 | 255.9 | 175.0 | 20 | 209.0 | 20 |
| Imidaclothiz | 3.17 | 30 | 262.0 | 180.8 | 15 | 122.0 | 26 |
| Acetamiprid | 3.17 | 45 | 223.0 | 125.9 | 18 | 55.9 | 12 |
| Pirimicarb | 3.36 | 40 | 239.0 | 72.0 | 20 | 182.0 | 16 |
| Thiacloprid | 3.58 | 45 | 253.0 | 125.9 | 20 | 217.0 | 12 |
| Aldicarb | 3.69 | 35 | 213.0 | 89.0 | 16 | 116.0 | 12 |
| Metolcarb | 3.96 | 25 | 164.0 | 109.0 | 12 | 94.0 | 26 |
| Propoxur | 4.21 | 5 | 210.0 | 111.0 | 15 | 168.0 | 5 |
| Carbofuran | 4.27 | 30 | 222.0 | 123.0 | 22 | 165.0 | 12 |
| Carbaryl | 4.43 | 25 | 202.0 | 127.0 | 25 | 145.0 | 10 |
| Isoprocarb | 4.72 | 30 | 194.0 | 95.0 | 15 | 137.0 | 9 |
| Promecarb | 5.25 | 30 | 208.0 | 109.0 | 17 | 151.0 | 9 |
| Analytes | Linear Range (μg/kg) | Regression Equation | R2 | LOD (μg/kg) | LOQ (μg/kg) | ME (%) | Fortification (μg/kg) | Recovery (%) | RSDr (%) | RSDR (%) |
|---|---|---|---|---|---|---|---|---|---|---|
| y = 11,641x – 21,927 | 1.0 | 92.2 | 6.9 | 1.8 | ||||||
| Propamocarb | 1.0–500.0 | 0.9998 | 0.2 | 1.0 | −3.4 | 10.0 | 88.7 | 4.5 | 7.0 | |
| 100.0 | 86.3 | 1.7 | 2.2 | |||||||
| y = 1026.8x − 258.23 | 10.0 | 97.9 | 5.7 | 3.7 | ||||||
| Dinotefuran | 10.0–500.0 | 0.9994 | 4.0 | 10.0 | −14.2 | 100.0 | 97.7 | 1.5 | 2.1 | |
| 500.0 | 96.7 | 1.1 | 1.9 | |||||||
| Oxamyl | 10.0–500.0 | y = 423.49x − 1011.8 | −14.8 | 10.0 | 104.0 | 7.8 | 8.3 | |||
| Oxamyl | 10.0–500.0 | 0.9997 | 4.0 | 10.0 | −14.8 | 100.0 | 98.6 | 3.5 | 2.6 | |
| 500.0 | 97.2 | 1.3 | 3.9 | |||||||
| y = 1710.8x − 3614.8 | 10.0 | 83.7 | 6.8 | 11.2 | ||||||
| Nitenpyram | 5.0–500.0 | 0.9997 | 2.0 | 5.0 | −3.3 | 100.0 | 86.3 | 1.6 | 3.1 | |
| 500.0 | 85.4 | 1.4 | 3.6 | |||||||
| y = 31,584x + 25,868 | 1.0 | 78.7 | 4.9 | 5.3 | ||||||
| Cycloxaprid | 1.0–100.0 | 0.9993 | 0.2 | 1.0 | 16.1 | 10.0 | 81.6 | 3.9 | 5.9 | |
| 100.0 | 82.1 | 1.0 | 5.3 | |||||||
| y = 3048.3x + 21,677 | 1.0 | 99.4 | 7.1 | 4.8 | ||||||
| Thiamethoxam | 1.0–500.0 | 0.9998 | 0.2 | 1.0 | 2.8 | 10.0 | 98.2 | 3.3 | 1.9 | |
| 100.0 | 99.1 | 1.5 | 1.2 | |||||||
| y = 787.1x + 1156.7 | 10.0 | 93.8 | 7.3 | 6.3 | ||||||
| Clothianidin | 1.0–500.0 | 0.9998 | 4.0 | 10.0 | −16.5 | 100.0 | 96.3 | 2.4 | 4.0 | |
| 500.0 | 97.8 | 2.9 | 2.7 | |||||||
| y = 1655.5x + 2906.1 | 1.0 | 97.7 | 4.0 | 8.6 | ||||||
| Imidacloprid | 1.0–500.0 | 0.9990 | 0.3 | 1.0 | 4.8 | 10.0 | 97.4 | 3.5 | 3.9 | |
| 100.0 | 97.9 | 0.9 | 2.2 | |||||||
| y = 1554.4x −4607.2 | 10.0 | 104.4 | 0.4 | 4.1 | ||||||
| Imidaclothiz | 10.0–500.0 | 0.9995 | 3.0 | 10.0 | 9.5 | 100.0 | 99.0 | 2.7 | 3.0 | |
| 500.0 | 98.7 | 0.9 | 2.4 | |||||||
| y = 15,870x + 17,493 | 1.0 | 92.1 | 4.0 | 3.0 | ||||||
| Acetamiprid | 1.0–200.0 | 1.0000 | 0.3 | 1.0 | 6.2 | 10.0 | 99.1 | 1.7 | 2.4 | |
| 100.0 | 99.6 | 0.8 | 0.9 | |||||||
| Pirimicarb | 1.0–500.0 | y = 16,692x + 30,784 | 0.9998 | 1.0 | −0.6 | 1.0 | 93.4 | 0.4 | 8.8 | |
| 1.0–500.0 | 0.9998 | 0.3 | 1.0 | −0.6 | 10.0 | 98.7 | 1.2 | 0.7 | ||
| 100.0 | 97.1 | 1.3 | 1.4 | |||||||
| ThiaclopridTHI | y = 22,576x + 21,938 | 1.0 | 99.8 | 1.7 | 1.0 | |||||
| 1.0–200.0 | 1.00000 | 0.2 | 1.0 | 3.7 | 10.0 | 100.5 | 1.0 | 2.0 | ||
| 100.0 | 99.3 | 1.4 | 1.3 | |||||||
| y = 1200.4x − 3213.9 | 10.0 | 102.9 | 4.2 | 7.3 | ||||||
| Aldicarb | 10.0–500.0 | 0.9995 | 3.0 | 10.0 | −16.1 | 100.0 | 99.6 | 1.7 | 0.9 | |
| 500.0 | 100.6 | 0.6 | 0.9 | |||||||
| y = 242.56x − 308.51 | 10.0 | 98.6 | 6.3 | 14.2 | ||||||
| Metolcarb | 10.0–1000.0 | 0.9992 | 3.0 | 10.0 | 9.4 | 100.0 | 96.6 | 3.3 | 1.2 | |
| 500.0 | 97.9 | 3.8 | 2.3 | |||||||
| y = 1524.9x − 598.33 | 10.0 | 103.6 | 3.9 | 1.4 | ||||||
| Propoxur | 5.0–500.0 | 0.9996 | 2.0 | 5.0 | −6.5 | 100.0 | 99.8 | 2.5 | 1.5 | |
| 500.0 | 99.8 | 0.9 | 1.8 | |||||||
| y = 22,528x + 10,821 | 1.0 | 86.5 | 1.9 | 4.3 | ||||||
| Carbofuran | 1.0–200.0 | 1.0000 | 0.3 | 1.0 | 3.8 | 10.0 | 102.9 | 2.7 | 2.8 | |
| 100.0 | 101.2 | 1.7 | 0.8 | |||||||
| y = 1192.2x − 166.83 | 10.0 | 105.7 | 4.4 | 6.3 | ||||||
| Carbaryl | 10.0–500.0 | 0.9997 | 3.0 | 10.0 | −7.4 | 100.0 | 96.9 | 3.1 | 2.4 | |
| 500.0 | 98.2 | 1.5 | 3.7 | |||||||
| y = 2748.7x + 6693.7 | 10.0 | 101.9 | 3.9 | 3.4 | ||||||
| Isoprocarb | 10.0–1000.0 | 0.9996 | 3.0 | 10.0 | −2.2 | 100.0 | 101.4 | 2.3 | 2.3 | |
| 500.0 | 101.0 | 2.5 | 2.1 | |||||||
| y = 3294.2x + 13,084 | 10.0 | 114.0 | 2.1 | 2.0 | ||||||
| Promecarb | 2.0–1000.0 | 0.9997 | 0.6 | 2.0 | −9.4 | 100.0 | 108.5 | 2.4 | 7.1 | |
| 500.0 | 102.8 | 2.1 | 2.1 |
| Compounds | Sample 1 (μg/kg) | Sample 2 (μg/kg) | Sample 3 (μg/kg) | Sample 4 (μg/kg) | Sample 5 (μg/kg) | Sample 6 (μg/kg) | Sample 7 (μg/kg) |
|---|---|---|---|---|---|---|---|
| Propamocarb | <LOD | <LOD | <LOD | 16.7 ± 1.1 | 15.5 ± 1.2 | 35.7 ± 2.1 | <LOD |
| Dinotefuran | <LOD | <LOD | <LOD | <LOD | <LOD | <LOD | <LOD |
| Oxamyl | <LOD | <LOD | <LOD | <LOD | <LOD | <LOD | <LOD |
| Nitenpyram | <LOD | <LOD | <LOD | <LOD | <LOD | <LOD | <LOD |
| Cycloxaprid | <LOD | <LOD | <LOD | <LOD | <LOD | <LOD | <LOD |
| Thiamethoxam | <LOD | <LOD | <LOD | <LOD | <LOD | <LOD | <LOD |
| Clothianidin | <LOD | <LOD | <LOD | <LOD | <LOD | <LOD | <LOD |
| Imidacloprid | <LOD | <LOD | <LOD | <LOD | <LOD | <LOD | 78.6 ± 6.0 |
| Imidaclothiz | <LOD | <LOD | <LOD | <LOD | <LOD | <LOD | <LOD |
| Acetamiprid | <LOD | <LOD | <LOD | <LOD | <LOD | <LOD | 4.0 ± 0.1 |
| Pirimicarb | <LOD | <LOD | <LOD | <LOD | <LOD | <LOD | <LOD |
| Thiacloprid | 4.5 ± 0.2 | 5.7 ± 0.2 | <LOD | 17.1 ± 0.5 | 5.0 ± 0.3 | <LOD | 6.9 ± 0.5 |
| Aldicarb | <LOD | <LOD | <LOD | <LOD | <LOD | <LOD | <LOD |
| Metolcarb | <LOD | <LOD | <LOD | <LOD | <LOD | <LOD | <LOD |
| Propoxur | <LOD | <LOD | <LOD | <LOD | <LOD | <LOD | <LOD |
| Carbofuran | <LOD | <LOD | <LOD | <LOD | <LOD | <LOD | <LOD |
| Carbaryl | <LOD | <LOD | <LOD | <LOD | <LOD | <LOD | <LOD |
| Isoprocarb | <LOD | <LOD | <LOD | <LOD | <LOD | <LOD | <LOD |
| Promecarb | <LOD | <LOD | <LOD | <LOD | <LOD | <LOD | <LOD |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, B.; Wang, S.; Ma, W.; Li, G.; Tu, M.; Ma, Z.; Zhang, Q.; Li, H.; Li, X. Simultaneous Determination of Neonicotinoid and Carbamate Pesticides in Freeze-Dried Cabbage by Modified QuEChERS and Ultra-Performance Liquid Chromatography–Tandem Mass Spectrometry. Foods 2023, 12, 699. https://doi.org/10.3390/foods12040699
Yang B, Wang S, Ma W, Li G, Tu M, Ma Z, Zhang Q, Li H, Li X. Simultaneous Determination of Neonicotinoid and Carbamate Pesticides in Freeze-Dried Cabbage by Modified QuEChERS and Ultra-Performance Liquid Chromatography–Tandem Mass Spectrometry. Foods. 2023; 12(4):699. https://doi.org/10.3390/foods12040699
Chicago/Turabian StyleYang, Bingxin, Sheng Wang, Wen Ma, Guanlin Li, Mengling Tu, Zhiyong Ma, Qinghe Zhang, Hongmei Li, and Xianjiang Li. 2023. "Simultaneous Determination of Neonicotinoid and Carbamate Pesticides in Freeze-Dried Cabbage by Modified QuEChERS and Ultra-Performance Liquid Chromatography–Tandem Mass Spectrometry" Foods 12, no. 4: 699. https://doi.org/10.3390/foods12040699