

Microbial Quality of Donkey Milk during Lactation Stages


Abstract
:1. Introduction
2. Materials and Methods
2.1. Collection and Preparation of DM
2.2. Sequencing
2.2.1. DNA Extraction
2.2.2. Amplification of 16S rRNA and Detection of PCR Products [16]
2.2.3. Library Preparation and Sequencing
2.3. Data Analysis
2.3.1. Paired-End Reads Merged and Quality Control
2.3.2. ASVs Denoise and Species Annotation
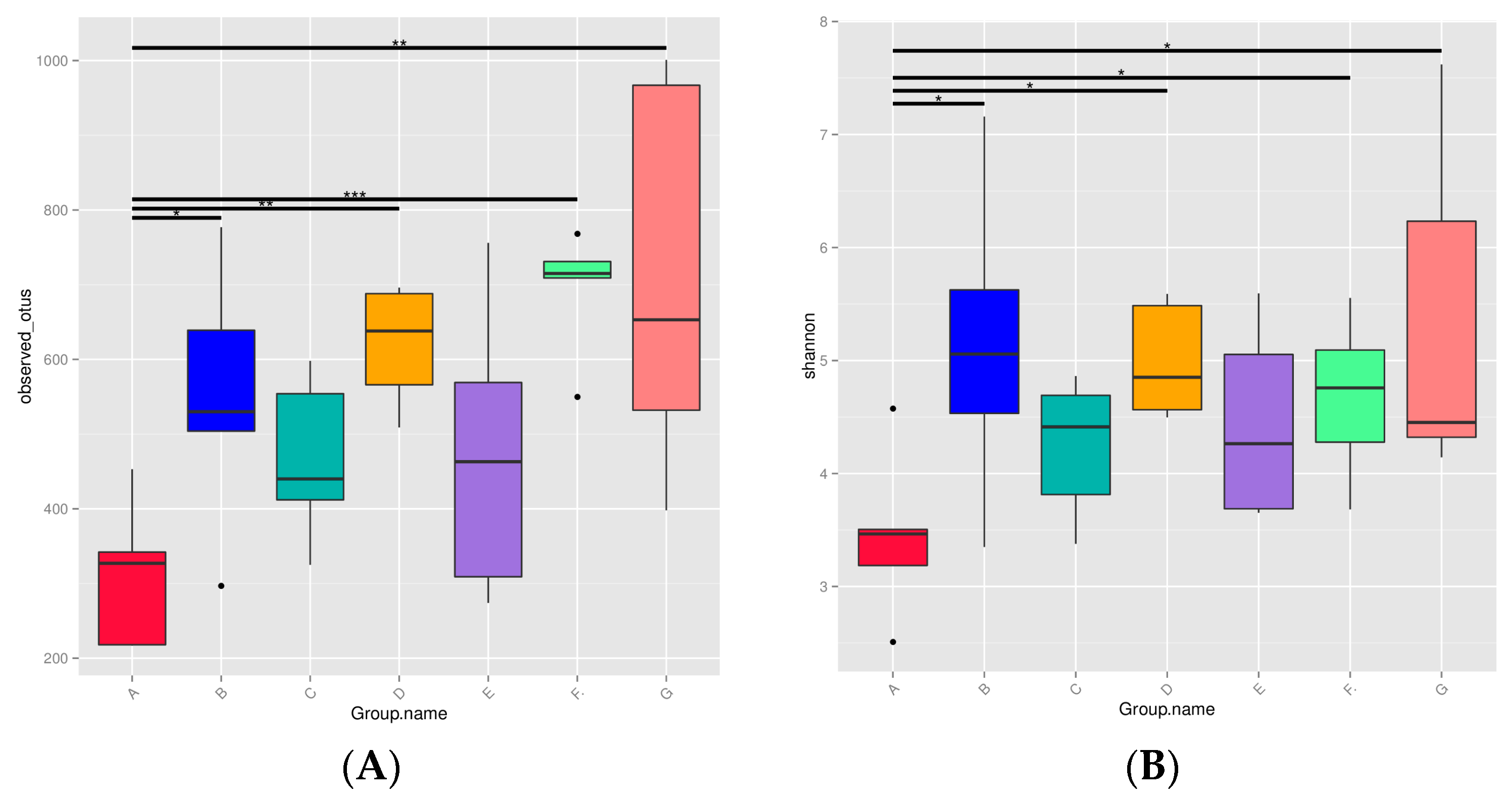
2.3.3. Alpha Diversity
2.3.4. Beta Diversity
2.3.5. Statistical Analysis
3. Results and Discussion
3.1. Diversity of Microbiota in DM from Different Lactation Stages
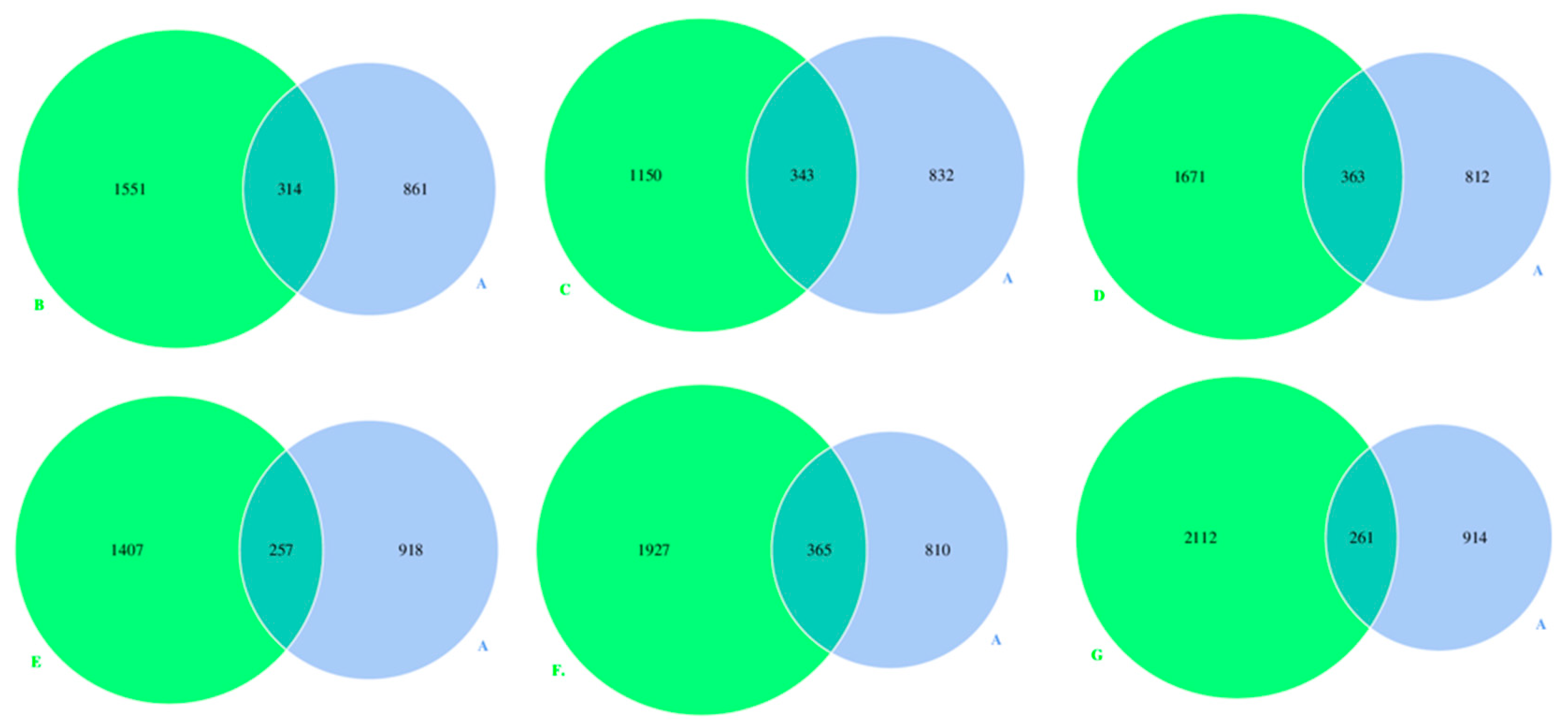
3.2. Microbes Numbers in DM of Different Lactation Stages
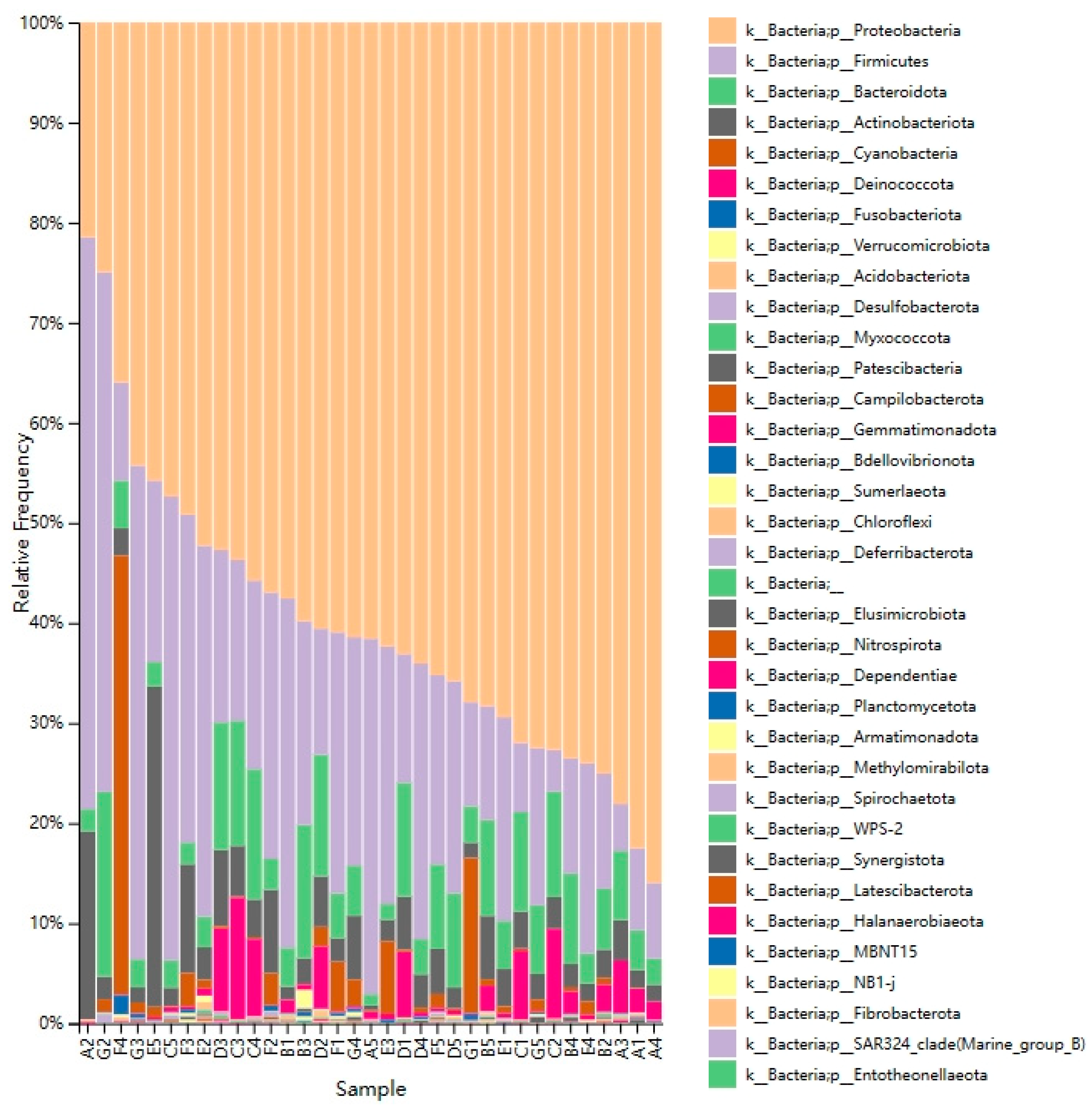
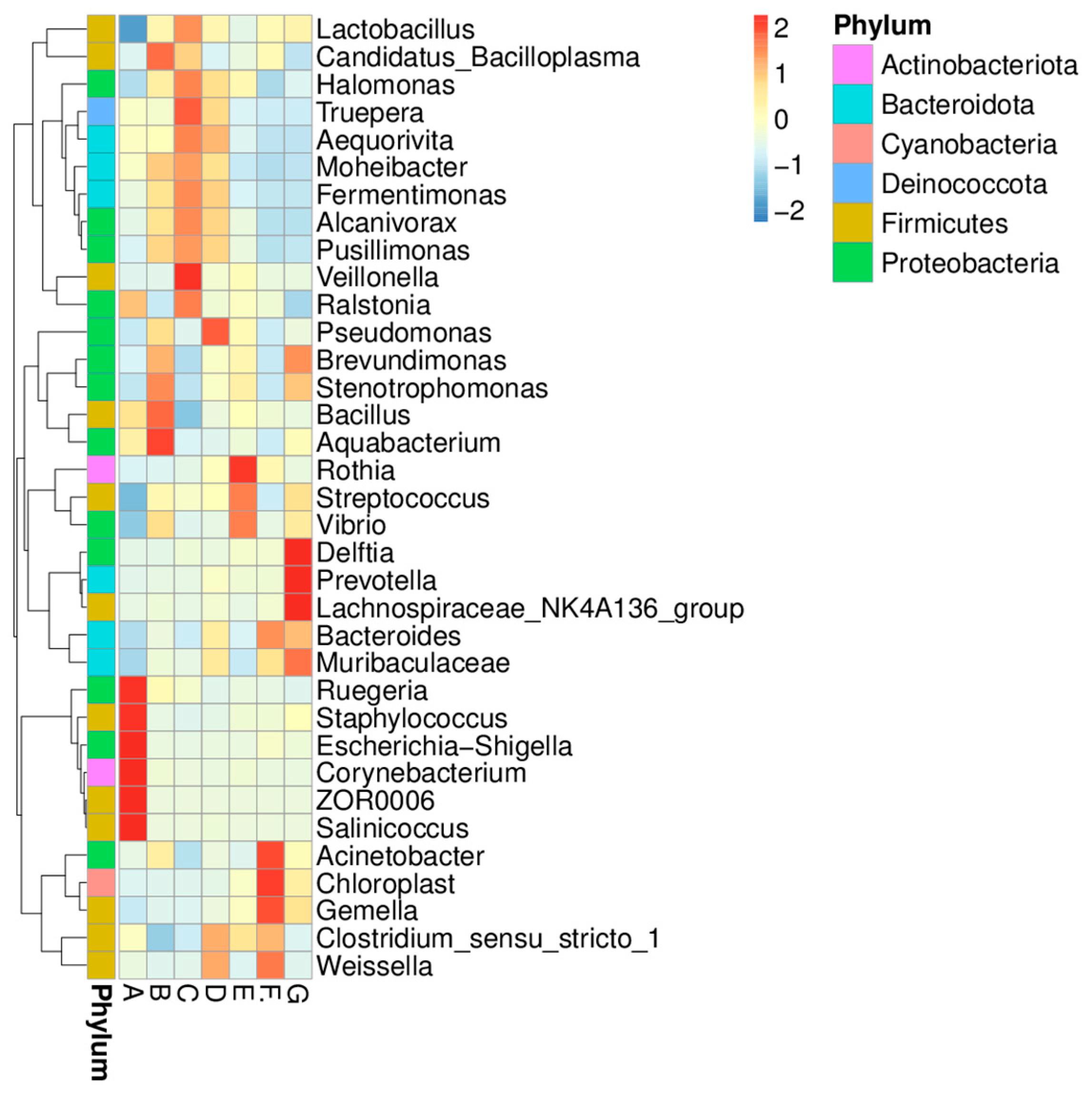
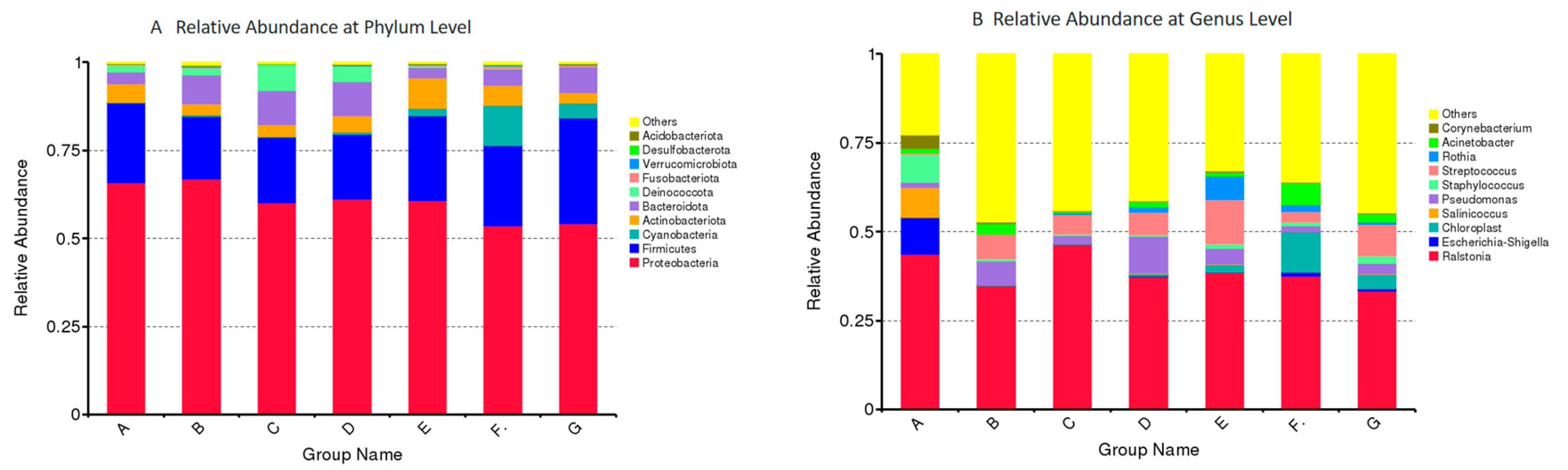
3.3. Microbiota Composition in DM of Different Lactation Stage
3.4. Functional Differences of Microbiome in DM of Different Lactation Stages
4. Discussion
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Guo, H.Y.; Pang, K.; Zhang, X.Y.; Zhao, L.; Chen, S.W.; Dong, M.L.; Ren, F.Z. Composition, Physiochemical properties, nitrogen fraction distribution, and amino acid profile of donkey milk. J. Dairy Sci. 2007, 90, 1635–1643. [Google Scholar] [CrossRef]
- Aspri, M.; Leni, G.; Galaverna, G.; Papademas, P. Bioactive properties of fermented donkey milk, before and after in vitro simulated gastrointestinal digestion. Food Chem. 2018, 268, 476–484. [Google Scholar] [CrossRef]
- Spada, V.; Ferranti, P.; Chianese, L.; Salimei, E.; Addeo, F.; Picariello, G. Antibacterial potential of donkey’s milk disclosed by untargeted proteomics. J. Proteom. 2021, 231, 104007. [Google Scholar] [CrossRef] [PubMed]
- Papademas, P.; Kamilari, E.; Aspri, M.; Anagnostopoulos, D.A.; Mousikos, P.; Kamilaris, A.; Tsaltas, D. Investigation of donkey milk bacterial diversity by 16S rDNA high-throughput sequencing on a Cyprus donkey farm. J. Dairy Sci. 2021, 104, 167–178. [Google Scholar] [CrossRef]
- Luo, J.; Jian, S.; Wang, P.; Ren, F.; Wang, F.; Chen, S. Thermal instability and characteristics of donkey casein micelles. Food Res. Int. 2019, 119, 436–443. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Fan, Y.; Shaikh, A.S.; Wang, Z.; Wang, D.; Tan, H. Dezhou donkey (Equus asinus) milk a potential treatment strategy for type 2 diabetes. J. Ethnopharmacol. 2020, 246, 112221. [Google Scholar] [CrossRef]
- Li, Q.; Li, M.; Zhang, J.; Shi, X.; Yang, M.; Zheng, Y.; Cao, X.; Yue, X.; Ma, S. Donkey milk inhibits triple-negative breast tumor progression and is associated with increased cleaved-caspase-3 expression. Food Funct. 2020, 11, 3053. [Google Scholar] [CrossRef] [PubMed]
- Kocic, H.; Langerholc, T.; Kostic, M.; Stojanovic, S.; Najman, S.; Krstic, M.; Nesic, I.; Godic, A.; Wollina, U. The regenerative potential of donkey and human milk on the redox-sensitive and proliferative signaling pathways of skin fibroblasts. Oxid. Med. Cell. Longev. 2020, 2020, 5618127. [Google Scholar] [CrossRef] [PubMed]
- Colavita, G.; Amadoro, C.; Rossi, F.; Fantuz, F.; Salimei, E. Hygienic characteristics and microbiological hazard identification in horse and donkey raw milk. Vet. Ital. 2016, 52, 21–29. [Google Scholar]
- Soto Del Rio, M.L.; Andrighetto, C.; Dalmasso, A.; Lombardi, A.; Civera, T.; Bottero, M.T. Isolation and characterisation of lactic acid bacteria from donkey milk. J. Dairy Res. 2016, 83, 383–386. [Google Scholar] [CrossRef]
- Derdak, R.; Quinteiro, J.; Sakoui, S.; Addoum, B.; Rodríguez Castro, J.; Rey Méndez, M.; Soukri, A.; El Khalfi, B. Isolation and identification of dominant bacteria from raw donkey milk produced in a region of Morocco by QIIME 2 and evaluation of their antibacterial activity. Sci. World J. 2021, 2021, 6664636. [Google Scholar] [CrossRef]
- Soto Del Rio, M.L.; Dalmasso, A.; Civera, T.; Bottero, M.T. Characterization of bacterial communities of donkey milk by high-throughput sequencing. Int. J. Food Microbiol. 2017, 251, 67–72. [Google Scholar] [CrossRef]
- Luoyizha, W.; Wu, X.; Zhang, M.; Guo, X.; Li, H.; Liao, X. Compared analysis of microbial diversity in donkey milk from Xinjiang and Shandong of China through High-throughput sequencing. Food Res. Int. 2020, 137, 109684. [Google Scholar] [CrossRef] [PubMed]
- Niamah, A.K. Detection of Listeria monocytogenes Bacteria in Four Types of Milk Using PCR. Pak. J. Nutr. 2012, 11, 1158–1160. [Google Scholar] [CrossRef]
- Wei, C.; Zhong, J.; Hu, T.; Zhao, X. Simultaneous detection of Escherichia coli O157: H7, Staphylococcus aureus and Salmonella by multiplex PCR in milk. 3 Biotech 2018, 8, 76. [Google Scholar] [CrossRef] [PubMed]
- Niyazbekova, Z.; Yao, X.T.; Liu, M.J.; Bold, N.; Tong, J.Z.; Chang, J.J.; Wen, Y.; Li, L.; Wang, Y.; Chen, D.K.; et al. Compositional and functional comparisons of the microbiota in the colostrum and mature milk of dairy goats. Animals 2020, 10, 1955. [Google Scholar] [CrossRef]
- Magoč, T.; Salzberg, S.L. FLASH: Fast length adjustment of short reads to improvegenome assemblies. Bioinformatics 2011, 21, 2957–2963. [Google Scholar] [CrossRef] [PubMed]
- Haas, B.J.; Gevers, D.; Earl, A.M.; Feldgarden, M.; Ward, D.V.; Giannoukos, G.; Ciulla, D.; Tabbaa, D.; Highlander, S.K.; Sodergren, E.; et al. Chimeric 16S rRNA sequence formation and detection in Sanger and 454-pyrosequenced PCR amplicons. Genome Res. 2011, 21, 494–504. [Google Scholar] [CrossRef]
- Li, M.; Shao, D.; Zhou, J.; Gu, J.; Qin, J.; Chen, W.; Wei, W. Signatures within esophageal microbiota with progression of esophageal squamous cell carcinoma. Chin. J. Cancer Res. 2020, 32, 755–767. [Google Scholar] [CrossRef]
- Li, B. Characterization of tetracycline resistant bacterial community in saline activated sludge using batch stress incubation with high-throughput sequencing analysis. Water Res. 2013, 47, 4207–4216. [Google Scholar] [CrossRef]
- Lozupone, C.; Knight, R. UniFrac: A new phylogenetic method for comparing microbial communities. Appl. Environ. Microbiol. 2005, 71, 8228–8235. [Google Scholar] [CrossRef] [PubMed]
- Lozupone, C. UniFrac: An effective distance metric for microbial community comparison. ISME J. 2011, 5, 169. [Google Scholar] [CrossRef]
- Lozupone, C. Quantitative and qualitative β diversity measures lead to different insights into factors that structure microbial communities. Appl. Environ. Microbiol. 2007, 73, 1576–1585. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Ma, Q.; Liu, G.; Wang, C. Effects of donkey milk on oxidative stress and inflammatory response. J. Food Biochem. 2022, 46, e13935. [Google Scholar] [CrossRef] [PubMed]
- Fantuz, F.; Ferraro, S.; Todini, L.; Cimarelli, L.; Fatica, A.; Marcantoni, F.; Salimei, E. Distribution of calcium, phosphorus, sulfur, magnesium, potassium, and sodium in major fractions of donkey milk. J. Dairy Sci. 2020, 103, 8741–8749. [Google Scholar] [CrossRef] [PubMed]
- Vincenzetti, S.; Santini, G.; Polzonetti, V.; Pucciarelli, S.; Klimanova, Y.; Polidori, P. Vitamins in human and donkey milk: Functional and nutritional role. Nutrients 2021, 13, 1509. [Google Scholar] [CrossRef]
- Trinchese, G.; Cavaliere, G.; Canani, R.B.; Matamoros, S.; Bergamo, P.; De Filippo, C.; Aceto, S.; Gaita, M.; Cerino, P.; Negri, R.; et al. Human, donkey and cow milk differently affects energy efficiency and inflammatory state by modulating mitochondrial function and gut microbiota. J. Nutr. Biochem. 2015, 26, 1136–1146. [Google Scholar] [CrossRef]
- De La Torre, U.; Henderson, J.D.; Furtado, K.L.; Pedroja, M.; Elenamarie, O.; Mora, A.; Pechanec, M.Y.; Maga, E.A.; Mienaltowski, M.J. Utilizing the fecal microbiota to understand foal gut transitions from birth to weaning. PLoS ONE 2019, 14, e0216211. [Google Scholar] [CrossRef]
- Martini, M.; Altomonte, I.; Salari, F.; Caroli, A.M. Short communication: Monitoring nutritional quality of Amiata donkey milk: Effects of lactation and productive season. J. Dairy Sci. 2014, 97, 6819–6822. [Google Scholar] [CrossRef] [PubMed]
- Raspa, F.; Cavallarin, L.; McLean, A.K.; Bergero, D.; Valle, E. A review of the appropriate nutrition welfare criteria of dairy donkeys: Nutritional requirements, farm management requirements and animal-based indicators. Animals 2019, 9, 315. [Google Scholar] [CrossRef]
- Li, M.; Li, W.; Wu, J.; Zheng, Y.; Shao, J.; Li, Q.; Kang, S.; Zhang, Z.; Yue, X.; Yang, M. Quantitative lipidomics reveals alterations in donkey milk lipids according to lactation. Food Chem. 2020, 310, 125866. [Google Scholar] [CrossRef] [PubMed]
- Lyons, K.E.; Shea, C.O.; Grimaud, G.; Ryan, C.A.; Dempsey, E.; Kelly, A.L.; Ross, R.P.; Stanton, C. The human milk microbiome aligns with lactation stage and not birth mode. Sci. Rep. 2022, 12, 5598. [Google Scholar] [CrossRef]
- Benson, K.F.; Carter, S.G.; Patterson, K.M.; Patel, D.; Jensen, G.S. A novel extract from bovine colostrum whey supports anti-bacterial and anti-viral innate immune functions in vitro and in vivo: I. Enhanced immune activity in vitro translates to improved microbial clearance in animal infection models. Prev. Med. 2012, 54, S116–S123. [Google Scholar] [CrossRef] [PubMed]
- Ward, T.L.; Hosid, S.; Ioshikhes, I.; Altosaar, I. Human milk metagenome: A functional capacity analysis. BMC. Microbiol. 2013, 13, 116. [Google Scholar] [CrossRef] [PubMed]
- Hooper, L.V.; Gordon, J.I. Commensal host-bacterial relationships in the gut. Science 2001, 292, 1115–1118. [Google Scholar] [CrossRef] [PubMed]
- Yvon, S.; Schwebel, L.; Belahcen, L.; Torm, H.; Peter, M.; Haimoud-Lekhal, D.A.; Eutamene, H.; Jard, G. Effects of thermized donkey milk with lysozyme activity on altered gut barrier in mice exposed to water-avoidance stress. J. Dairy Sci. 2019, 102, 7697–7706. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| KO_Hierarchy | KEGG_Description | p Value | |
|---|---|---|---|
| A vs. B | K02004 | putative ABC transport system permease protein | 0.02 |
| K02529 | LacI family transcriptional regulator | 0.021 | |
| K02495 | oxygen-independent coproporphyrinogen III oxidase (EC:1.3.98.3) | 0.038 | |
| K02342 | DNA polymerase III subunit epsilon (EC:2.7.7.7) | 0.030 | |
| A vs. C | K02003 | putative ABC transport system ATP-binding protein | 0.026 |
| K02004 | putative ABC transport system permease protein | 0.014 | |
| K02529 | LacI family transcriptional regulator | 0.028 | |
| K15634 | probable phosphoglycerate mutase (EC:5.4.2.12) | 0.012 | |
| K02342 | DNA polymerase III subunit epsilon (EC:2.7.7.7) | 0.025 | |
| A vs. D | K02003 | putative ABC transport system ATP-binding protein | 0.015 |
| K02004 | putative ABC transport system permease protein | 0.006 | |
| K02529 | LacI family transcriptional regulator | 0.004 | |
| K15634 | probable phosphoglycerate mutase (EC:5.4.2.12) | 0.011 | |
| K02342 | DNA polymerase III subunit epsilon (EC:2.7.7.7) | 0.036 | |
| A vs. E | K02003 | putative ABC transport system ATP-binding protein | 0.009 |
| K02004 | putative ABC transport system permease protein | 0.007 | |
| K02030 | polar amino acid transport system substrate-binding protein | 0.040 | |
| K03574 | 8-oxo-dGTP diphosphatase (EC:3.6.1.55) | 0.029 | |
| K15634 | probable phosphoglycerate mutase (EC:5.4.2.12) | 0.015 | |
| K01462 | peptide deformylase (EC:3.5.1.88) | 0.019 | |
| K03100 | signal peptidase I (EC:3.4.21.89) | 0.013 | |
| K02040 | phosphate transport system substrate-binding protein | 0.023 | |
| K02342 | dnaQ; DNA polymerase III subunit epsilon (EC:2.7.7.7) | 0.039 | |
| K07052 | uncharacterized protein | 0.005 | |
| A vs. F | K02004 | putative ABC transport system permease protein | 0.009 |
| K03657 | DNA helicase II / ATP-dependent DNA helicase PcrA (EC:3.6.4.12) | 0.037 | |
| K15634 | probable phosphoglycerate mutase (EC:5.4.2.12) | 0.038 | |
| K04487 | cysteine desulfurase (EC:2.8.1.7) | 0.005 | |
| K01462 | peptide deformylase (EC:3.5.1.88) | 0.029 | |
| K03100 | signal peptidase I (EC:3.4.21.89) | 0.013 | |
| A vs. G | K02004 | putative ABC transport system permease protein | 0.045 |
| K03704 | cold shock protein (beta-ribbon, CspA family) | 0.014 | |
| K15634 | probable phosphoglycerate mutase (EC:5.4.2.12) | 0.024 | |
| K02495 | oxygen-independent coproporphyrinogen III oxidase (EC:1.3.98.3) | 0.012 | |
| K01462 | peptide deformylase (EC:3.5.1.88) | 0.016 | |
| K03100 | signal peptidase I (EC:3.4.21.89) | 0.007 | |
| K07052 | uncharacterized protein | 0.030 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, M.; Huang, F.; Du, X.; Wang, C.; Liu, G. Microbial Quality of Donkey Milk during Lactation Stages. Foods 2023, 12, 4272. https://doi.org/10.3390/foods12234272
Zhou M, Huang F, Du X, Wang C, Liu G. Microbial Quality of Donkey Milk during Lactation Stages. Foods. 2023; 12(23):4272. https://doi.org/10.3390/foods12234272
Chicago/Turabian StyleZhou, Miaomiao, Fei Huang, Xinyi Du, Changfa Wang, and Guiqin Liu. 2023. "Microbial Quality of Donkey Milk during Lactation Stages" Foods 12, no. 23: 4272. https://doi.org/10.3390/foods12234272