
Redox Targets for Phosphine–Boranes

Abstract
:1. Introduction
2. Results
2.1. Global Effects of Borane-Containing and Non-Borane-Containing Reducing Drugs on the Cysteines of 661W Proteins
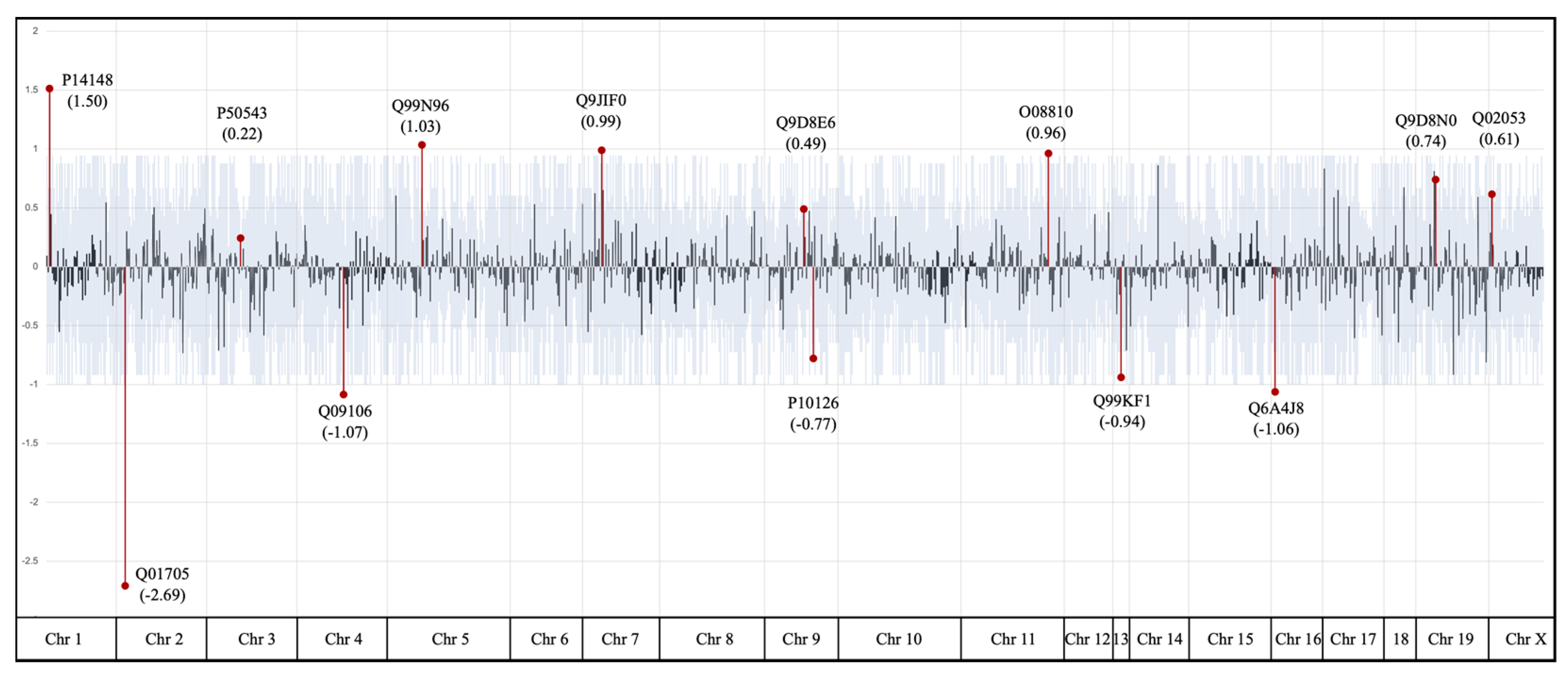
2.2. Effects of PB1 on the Cysteines Present in 661W Cell Proteins
2.3. Biological Relevance of Identified Targets
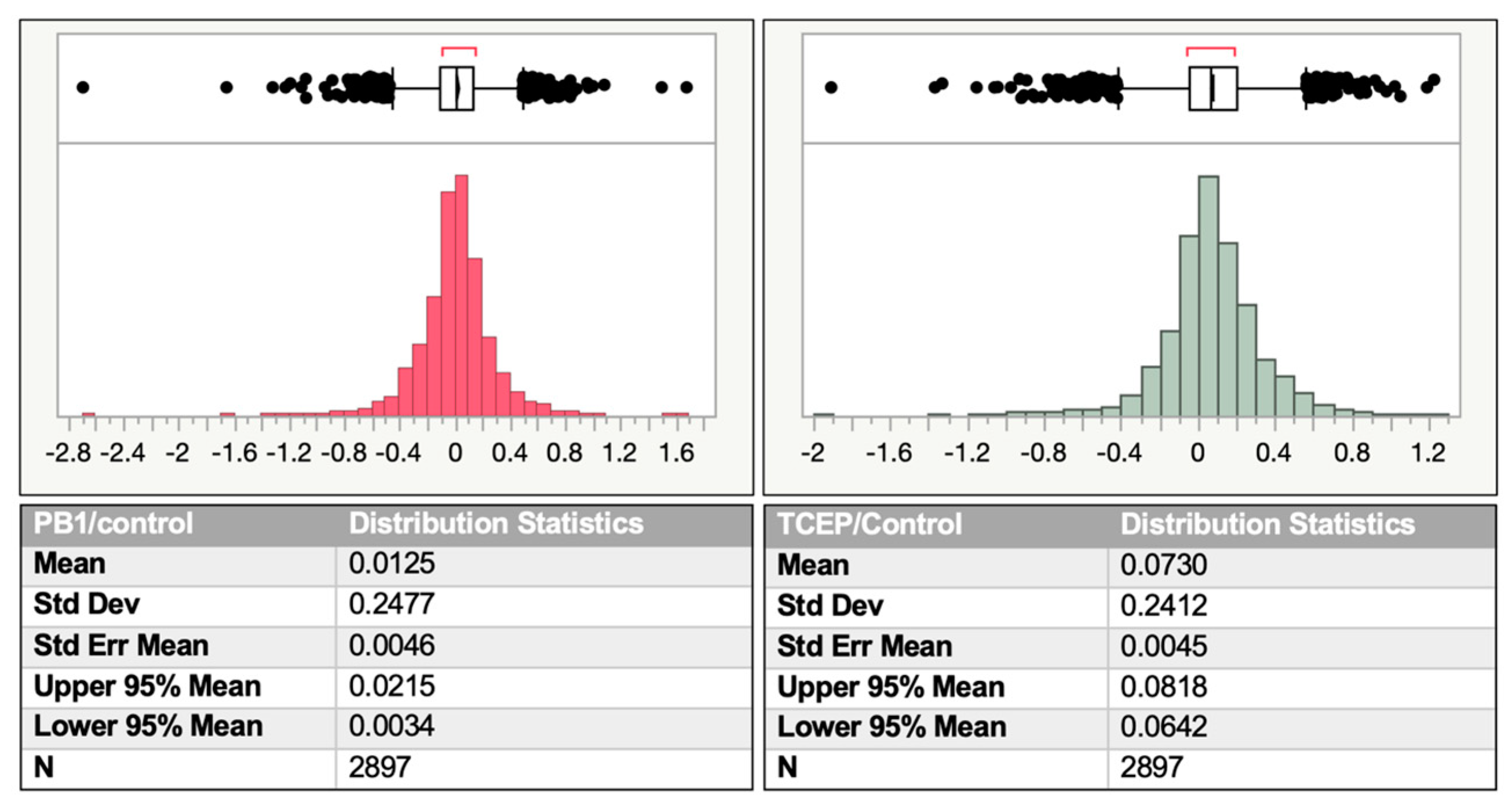
2.4. Comparison between PB1- and TCEP-Mediated Reduction
3. Discussion
4. Materials and Methods
4.1. Synthesis of PB1
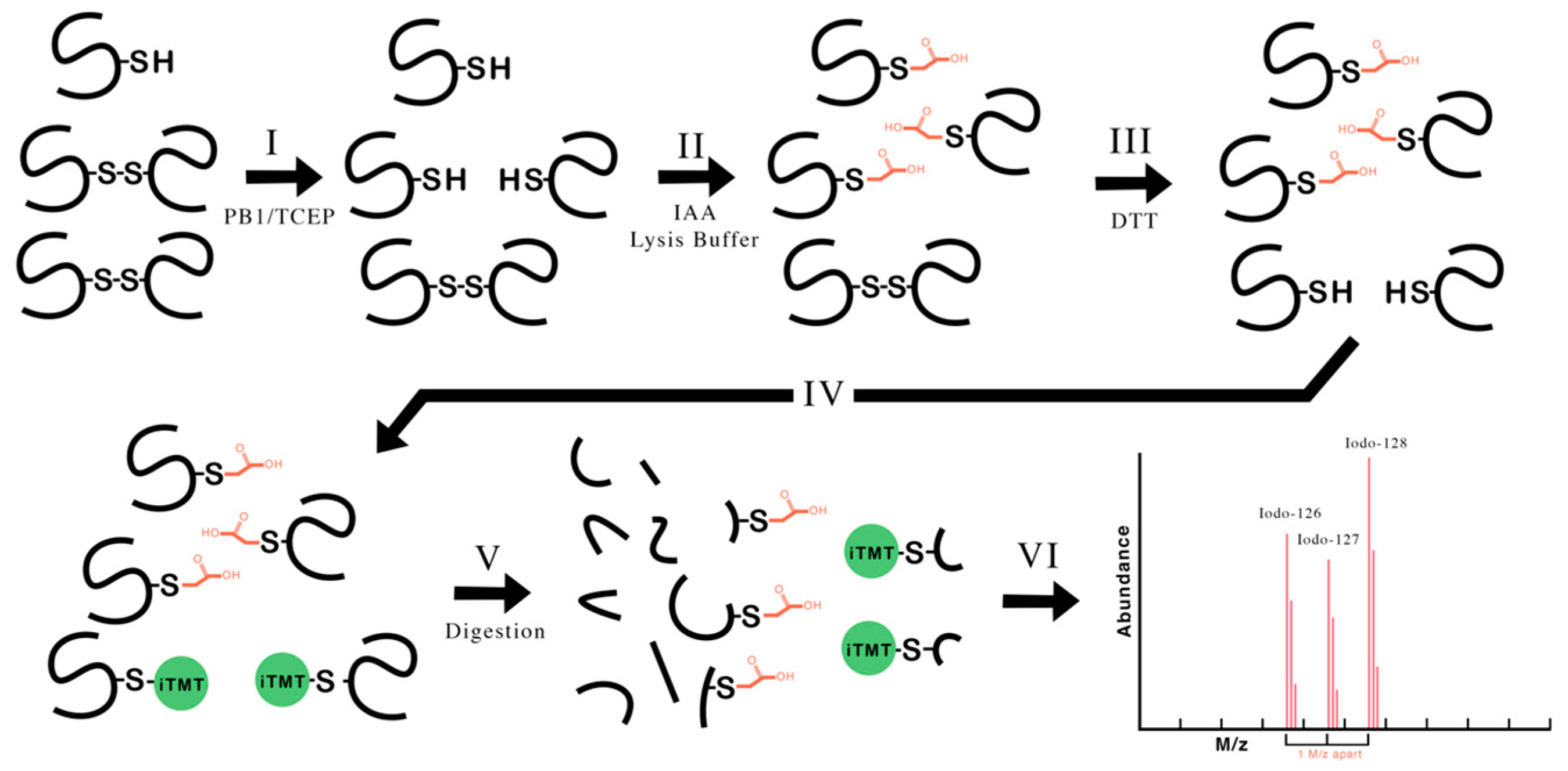
4.2. Protocol to Identify Disulfide Bonds Associated with Phosphine–Borane Complex Activity
4.3. Exposure of 661W Cells to Borane-Containing or Non-Borane-Containing Reducing Drugs
4.4. Cell Harvest Protocol
4.5. Labeling Protocol
4.6. Mass Spectrometer System and Operation
4.7. Mass Spectrometer Data Processing and Analysis
4.8. Statistics
4.9. Toxicity Assessment
4.10. Consistency of Parallel Cultures
4.11. Detection of iodoTMT Labeling
4.12. Prediction of Unknown Protein Structures
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Avdeeva, V.V.; Garaev, T.M.; Breslav, N.V.; Burtseva, E.I.; Grebennikova, T.V.; Zhdanov, A.P.; Zhizhin, K.Y.; Malinina, E.A.; Kuznetsov, N.T. New type of RNA virus replication inhibitor based on decahydro-closo-decaborate anion containing amino acid ester pendant group. JBIC J. Biol. Inorg. Chem. 2022, 27, 421–429. [Google Scholar] [CrossRef]
- Ali, F.; S Hosmane, N.; Zhu, Y. Boron chemistry for medical applications. Molecules 2020, 25, 828. [Google Scholar] [CrossRef] [Green Version]
- Fink, K.; Uchman, M. Boron cluster compounds as new chemical leads for antimicrobial therapy. Coord. Chem. Rev. 2021, 431, 213684. [Google Scholar] [CrossRef]
- Geiger, L.K.; Kortuem, K.R.; Alexejun, C.; Levin, L.A. Reduced redox state allows prolonged survival of axotomized neonatal retinal ganglion cells. Neuroscience 2002, 109, 635–642. [Google Scholar] [CrossRef] [PubMed]
- Schlieve, C.R.; Tam, A.; Nilsson, B.L.; Lieven, C.J.; Raines, R.T.; Levin, L.A. Synthesis and characterization of a novel class of reducing agents that are highly neuroprotective for retinal ganglion cells. Exp. Eye Res. 2006, 83, 1252–1259. [Google Scholar] [CrossRef] [PubMed]
- Swanson, K.I.; Schlieve, C.R.; Lieven, C.J.; Levin, L.A. Neuroprotective effect of sulfhydryl reduction in a rat optic nerve crush model. Investig. Ophthalmol. Vis. Sci. 2005, 46, 3737–3741. [Google Scholar] [CrossRef]
- Almasieh, M.; Lieven, C.J.; Levin, L.A.; Di Polo, A. A cell-permeable phosphine-borane complex delays retinal ganglion cell death after axonal injury through activation of the pro-survival extracellular signal-regulated kinases 1/2 pathway. J. Neurochem. 2011, 118, 1075–1086. [Google Scholar] [CrossRef]
- Dmitrenko, O.; Thorpe, C.; Bach, R.D. Mechanism of SN2 disulfide bond cleavage by phosphorus nucleophiles. Implications for biochemical disulfide reducing agents. J. Org. Chem. 2007, 72, 8298–8307. [Google Scholar] [CrossRef] [Green Version]
- Lloyd-Jones, G.C.; Taylor, N.P. Mechanism of phosphine borane deprotection with amines: The effects of phosphine, solvent and amine on rate and efficiency. Chem. A Eur. J. 2015, 21, 5423–5428. [Google Scholar] [CrossRef]
- Remtulla, R.; Das, S.K.; Levin, L.A. Predicting Absorption-Distribution Properties of Neuroprotective Phosphine-Borane Compounds Using In Silico Modeling and Machine Learning. Molecules 2021, 26, 2505. [Google Scholar] [CrossRef]
- Adler, V.; Yin, Z.; Tew, K.D.; Ronai, Z.E. Role of redox potential and reactive oxygen species in stress signaling. Oncogene 1999, 18, 6104–6111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niemuth, N.J.; Thompson, A.F.; Crowe, M.E.; Lieven, C.J.; Levin, L.A. Intracellular disulfide reduction by phosphine-borane complexes: Mechanism of action for neuroprotection. Neurochem. Int. 2016, 99, 24–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sen, C.K.; Packer, L. Antioxidant and redox regulation of gene transcription. FASEB J. 1996, 10, 709–720. [Google Scholar] [CrossRef] [PubMed]
- Dao, V.T.-V.; Casas, A.I.; Maghzal, G.J.; Seredenina, T.; Kaludercic, N.; Robledinos-Anton, N.; Di Lisa, F.; Stocker, R.; Ghezzi, P.; Jaquet, V. Pharmacology and clinical drug candidates in redox medicine. Antioxid. Redox Signal. 2015, 23, 1113–1129. [Google Scholar] [CrossRef] [Green Version]
- Tew, K.D.; Townsend, D.M. Redox platforms in cancer drug discovery and development. Curr. Opin. Chem. Biol. 2011, 15, 156–161. [Google Scholar] [CrossRef] [Green Version]
- Lieven, C.J.; Ribich, J.D.; Crowe, M.E.; Levin, L.A. Redox proteomic identification of visual arrestin dimerization in photoreceptor degeneration after photic injury. Investig. Ophthalmol. Vis. Sci. 2012, 53, 3990–3998. [Google Scholar] [CrossRef]
- Nicholson, B.; Kumar, K.S. The multifaceted roles of USP7: New therapeutic opportunities. Cell Biochem. Biophys. 2011, 60, 61–68. [Google Scholar] [CrossRef]
- Haas, A.L.; Warms, J.V.; Rose, I.A. Ubiquitin adenylate: Structure and role in ubiquitin activation. Biochemistry 1983, 22, 4388–4394. [Google Scholar] [CrossRef]
- Simões, V.; Harley, L.; Cizubu, B.K.; Zhou, Y.; Pajak, J.; Snyder, N.A.; Bouvette, J.; Borgnia, M.J.; Arya, G.; Bartesaghi, A.; et al. Redox sensitive E2 Rad6 controls cellular response to oxidative stress via K63 ubiquitination of ribosomes. bioRxiv 2021. [CrossRef]
- Doris, K.S.; Rumsby, E.L.; Morgan, B.A. Oxidative stress responses involve oxidation of a conserved ubiquitin pathway enzyme. Mol. Cell. Biol. 2012, 32, 4472–4481. [Google Scholar] [CrossRef] [Green Version]
- Gamalinda, M.; Ohmayer, U.; Jakovljevic, J.; Kumcuoglu, B.; Woolford, J.; Mbom, B.; Lin, L.; Woolford, J.L. A hierarchical model for assembly of eukaryotic 60S ribosomal subunit domains. Genes Dev. 2014, 28, 198–210. [Google Scholar] [CrossRef] [Green Version]
- Willi, J.; Küpfer, P.; Evéquoz, D.; Fernandez, G.; Katz, A.; Leumann, C.; Polacek, N. Oxidative stress damages rRNA inside the ribosome and differentially affects the catalytic center. Nucleic Acids Res. 2018, 46, 1945–1957. [Google Scholar] [CrossRef] [Green Version]
- Shakir, S.; Vinh, J.; Chiappetta, G. Quantitative analysis of the cysteine redoxome by iodoacetyl tandem mass tags. Anal. Bioanal. Chem. 2017, 409, 3821–3830. [Google Scholar] [CrossRef]
- Forman, H.J.; Zhang, H.; Rinna, A. Glutathione: Overview of its protective roles, measurement, and biosynthesis. Mol. Asp. Med. 2009, 30, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Huang, K.-P.; Huang, F.L. Glutathionylation of proteins by glutathione disulfide S-oxide. Biochem. Pharmacol. 2002, 64, 1049–1056. [Google Scholar] [CrossRef] [PubMed]
- Morgan, B.; Ezeriņa, D.; Amoako, T.N.; Riemer, J.; Seedorf, M.; Dick, T.P. Multiple glutathione disulfide removal pathways mediate cytosolic redox homeostasis. Nat. Chem. Biol. 2013, 9, 119–125. [Google Scholar] [CrossRef]
- Chakravarthi, S.; Bulleid, N.J. Glutathione is required to regulate the formation of native disulfide bonds within proteins entering the secretory pathway. J. Biol. Chem. 2004, 279, 39872–39879. [Google Scholar] [CrossRef] [Green Version]
- Almasieh, M.; Wilson, A.M.; Morquette, B.; Cueva Vargas, J.L.; Di Polo, A. The molecular basis of retinal ganglion cell death in glaucoma. Prog. Retin. Eye Res. 2012, 31, 152–181. [Google Scholar] [CrossRef] [PubMed]
- He, S.; Stankowska, D.L.; Ellis, D.Z.; Krishnamoorthy, R.R.; Yorio, T. Targets of neuroprotection in glaucoma. J. Ocul. Pharmacol. Ther. 2018, 34, 85–106. [Google Scholar] [CrossRef] [PubMed]
- Penke, B.; Bogár, F.; Crul, T.; Sántha, M.; Tóth, M.E.; Vígh, L. Heat shock proteins and autophagy pathways in neuroprotection: From molecular bases to pharmacological interventions. Int. J. Mol. Sci. 2018, 19, 325. [Google Scholar] [CrossRef] [Green Version]
- Wubben, T.J.; Zacks, D.N.; Besirli, C.G. Retinal neuroprotection: Current strategies and future directions. Curr. Opin. Ophthalmol. 2019, 30, 199–205. [Google Scholar] [CrossRef]
- Sriuranpong, V.; Borges, M.W.; Ravi, R.K.; Arnold, D.R.; Nelkin, B.D.; Baylin, S.B.; Ball, D.W. Notch signaling induces cell cycle arrest in small cell lung cancer cells. Cancer Res. 2001, 61, 3200–3205. [Google Scholar] [PubMed]
- Doyle, K.; Fitzpatrick, F. Redox signaling, alkylation (carbonylation) of conserved cysteines inactivates class I histone deacetylases 1, 2, and 3 and antagonizes their transcriptional repressor function. J. Biol. Chem. 2010, 285, 17417–17424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crowe, M.E.; Lieven, C.J.; Thompson, A.F.; Sheibani, N.; Levin, L.A. Borane-protected phosphines are redox-active radioprotective agents for endothelial cells. Redox Biol. 2015, 6, 73–79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nelson, P.R.; Wludyka, P.S.; Copeland, K.A. The Analysis of Means: A Graphical Method for Comparing Means, Rates, and Proportions; SIAM: Philadelphia, PA, USA, 2005. [Google Scholar]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Protein | Protein Accession Code | Annotated Sequence | Positions in Proteins | Abundance Ratio (Sample/Control) | p |
|---|---|---|---|---|---|
| Neurogenic locus notch homolog protein 1 | Q01705 | [R].CEGDVNEcLSNPcDPR.[G] | [1264–1279] | 0.068 | 5.0 × 10−11 |
| Thrombospondin-4 | Q9Z1T2 | [K].DGIGDEcDDDDDNDGIPDLVPPGPDNcR.[L] | [659–686] | 0.330 | 1.1 × 10−3 |
| Histone deacetylase 1 | O09106 | [R].FNVGEDcPVFDGLFEFcQLSTGGSVASAVK.[L] | [94–123] | 0.340 | 2.1 × 10−3 |
| Ubiquitin carboxyl-terminal hydrolase 7 | Q6A4J8 | [K].SFGWETLDSFMQHDVQELcR.[V] | [283–302] | 0.341 | 2.2 × 10−3 |
| Elongation factor 1-alpha 1 | P10126 | [K].NMITGTSQADcAVLIVAAGVGEFEAGISK.[N] | [101–129] | 0.491 | 2.8 × 10−6 |
| Transmembrane emp24 domain-containing protein 9 | Q99KF1 | [KR].cFIEEIPDETmVIGNYR.[T] | [49–65] | 0.887 | 3.4 × 10−2 |
| Protein S100-A11 | P50543 | [R].cIESLIAVFQK.[Y] | [8–18] | 1.307 | 2.9 × 10−2 |
| Elongation factor 1-gamma | Q9D8N0 | [R].WFLTcINQPQFR.[A] | [190–201] | 1.712 | 4.3 × 10−4 |
| Ubiquitin-like modifier-activating enzyme 1 | Q02053 | [K].IIPAIATTTAAVVGLVcLELYK.[V] | [890–911] | 1.873 | 5.3 × 10−3 |
| DNA dC->dU-editing enzyme APOBEC-3 | Q99J72 | [R].LYNVQDPETQQNLcR.[L] | [142–156] | 2.558 | 3.6 × 10−9 |
| 116 kDa U5 small nuclear ribonucleoprotein component | O08810 | [K].VEESGEHVILGTGELYLDcVMHDLR.[K] | [620–644] | 2.615 | 3.6 × 10−2 |
| 60S ribosomal protein L4 | Q9D8E6 | [R].FcIWTESAFR.[K] | [249–258] | 3.012 | 8.5 × 10−10 |
| 60S ribosomal protein L7 | P14148 | [K].FGIIcMEDLIHEIYTVGK.[R] | [204–221] | 4.484 | 1.0 × 10−7 |
| Protein | Protein Accession Code | Annotated Sequence | Positions in Proteins | Abundance Ratio (Sample/Control) | p |
|---|---|---|---|---|---|
| Neurogenic locus notch homolog protein 1 | Q01705 | [R].CEGDVNEcLSNPcDPR.[G] | [1264–1279] | 0.149 | 5.0 × 10−11 |
| Collagen alpha-1(I) chain | P11087 | [K].SGEYWIDPNQGcNLDAIK.[V] | [1260–1277] | 0.629 | 2.1 × 10−2 |
| Elongation factor 1-alpha 1 | P10126 | [K].NMITGTSQADcAVLIVAAGVGEFEAGISK] | [101–129] | 0.661 | 7.9 × 10−3 |
| Phosphoglycerate kinase 1 | P09411 | [K].TGQATVASGIPAGWMGLDcGTESSK.[K] | [298–322] | 0.736 | 8.5 × 10−3 |
| Elongation factor 2 | P58252 | [R].LmEPIYLVEIQcPEQVVGGIYGVLNR.[K] | [740–765] | 0.886 | 6.2 × 10−5 |
| Transmembrane emp24 domain-containing protein 9 | Q99KF1 | [KR].cFIEEIPDETmVIGNYR.[T] | [49–65] | 1.046 | 4.4 × 10−2 |
| DNA dC->dU-editing enzyme APOBEC-3 | Q99J72 | [R].LYNVQDPETQQNLcR.[L] | [142–156] | 2.278 | 10.0 × 10−6 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Morocz, Y.; Greben, R.E.; Levin, L.A. Redox Targets for Phosphine–Boranes. Inorganics 2023, 11, 310. https://doi.org/10.3390/inorganics11070310
Morocz Y, Greben RE, Levin LA. Redox Targets for Phosphine–Boranes. Inorganics. 2023; 11(7):310. https://doi.org/10.3390/inorganics11070310
Chicago/Turabian StyleMorocz, Yonatan, Rachel E. Greben, and Leonard A. Levin. 2023. "Redox Targets for Phosphine–Boranes" Inorganics 11, no. 7: 310. https://doi.org/10.3390/inorganics11070310