
Ligand Tuning in Cu(pyalk)2 Water Oxidation Electrocatalysis

 ,
,
Abstract
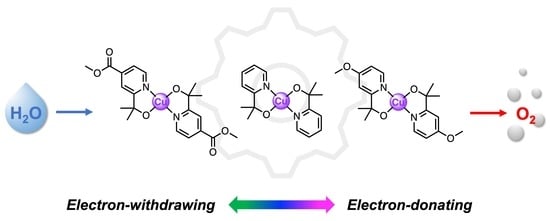
:
1. Introduction
2. Results and Discussion
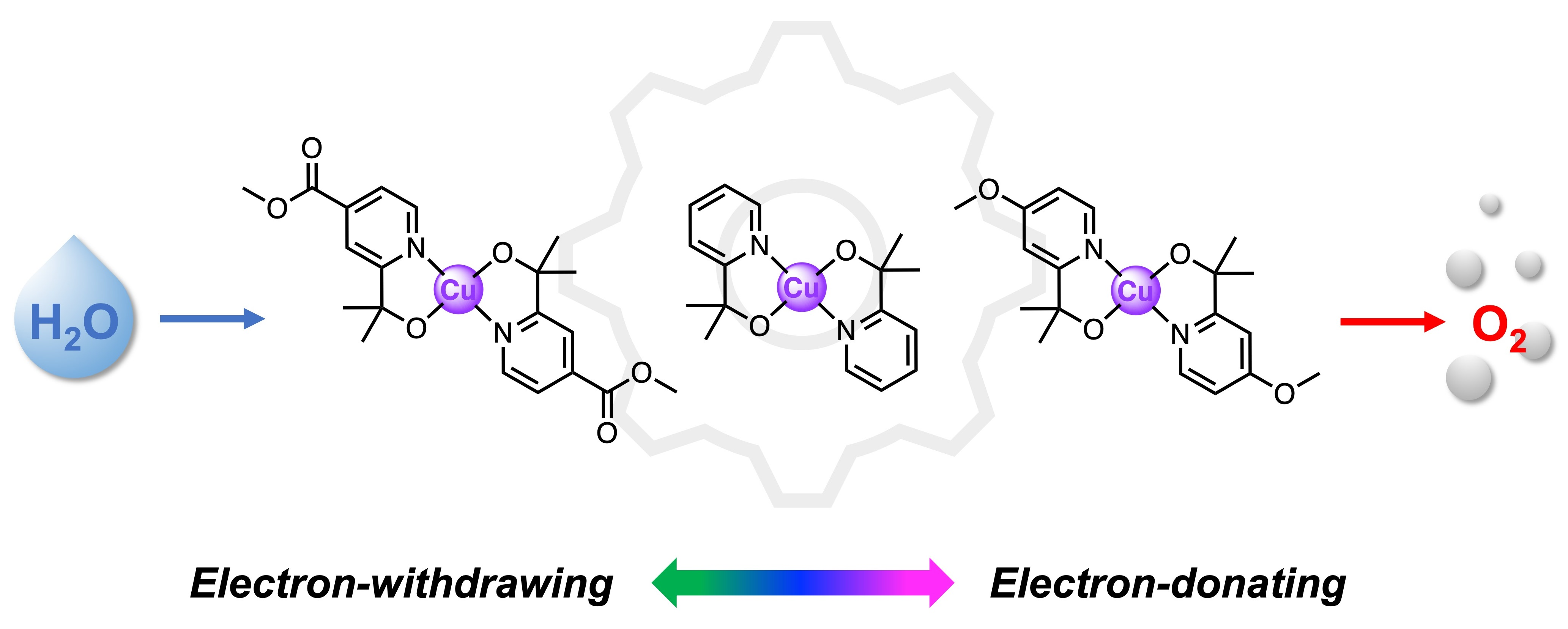
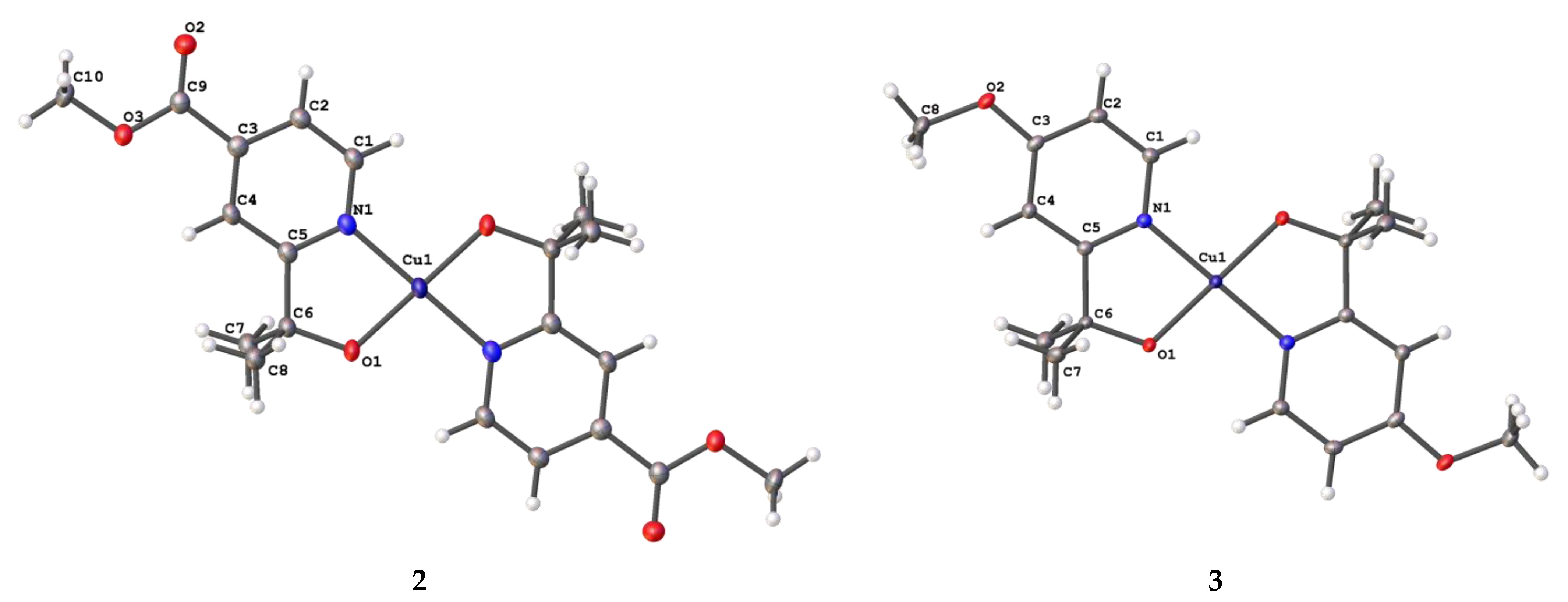
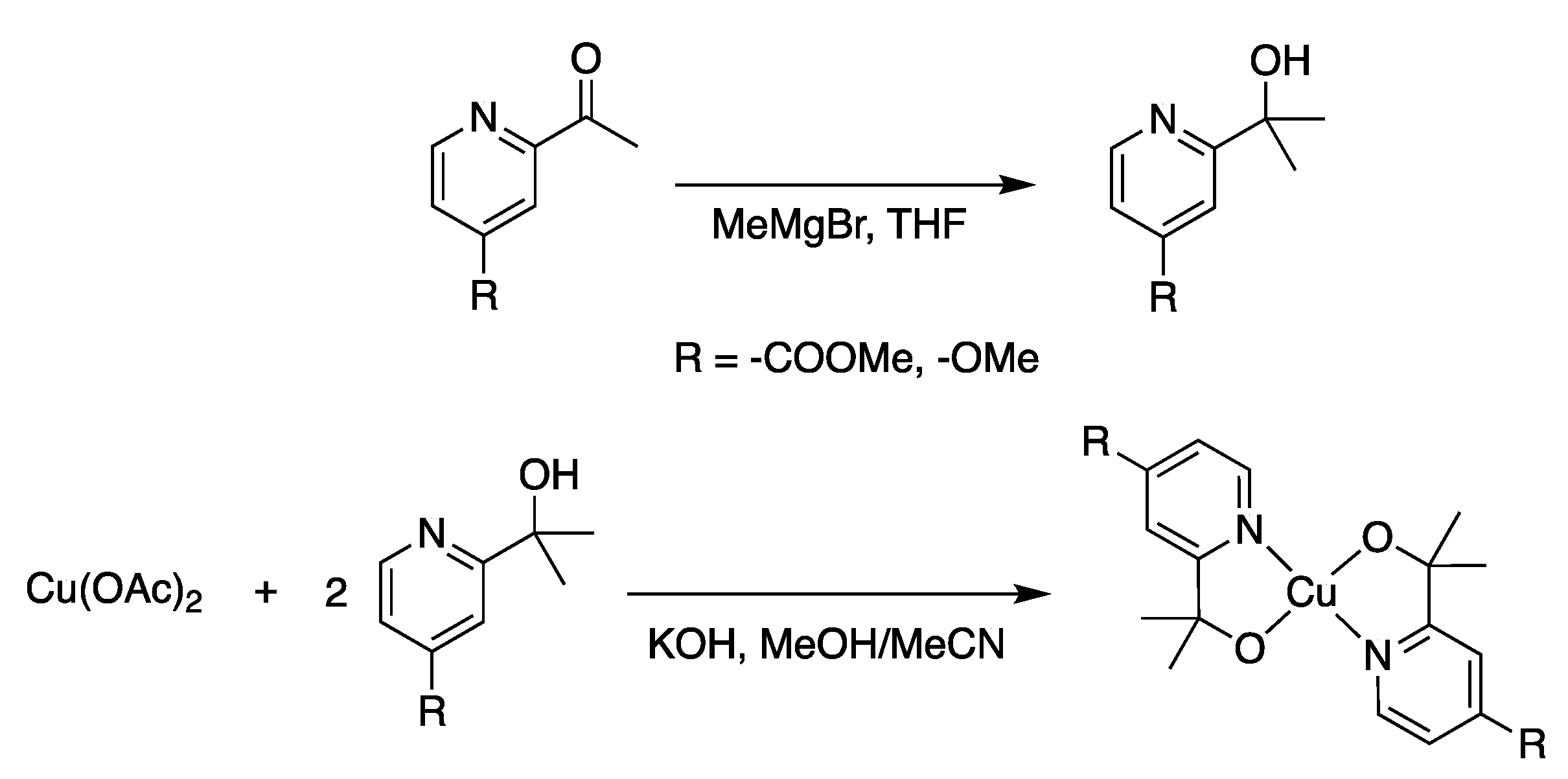
2.1. Synthesis and Structural Characterization
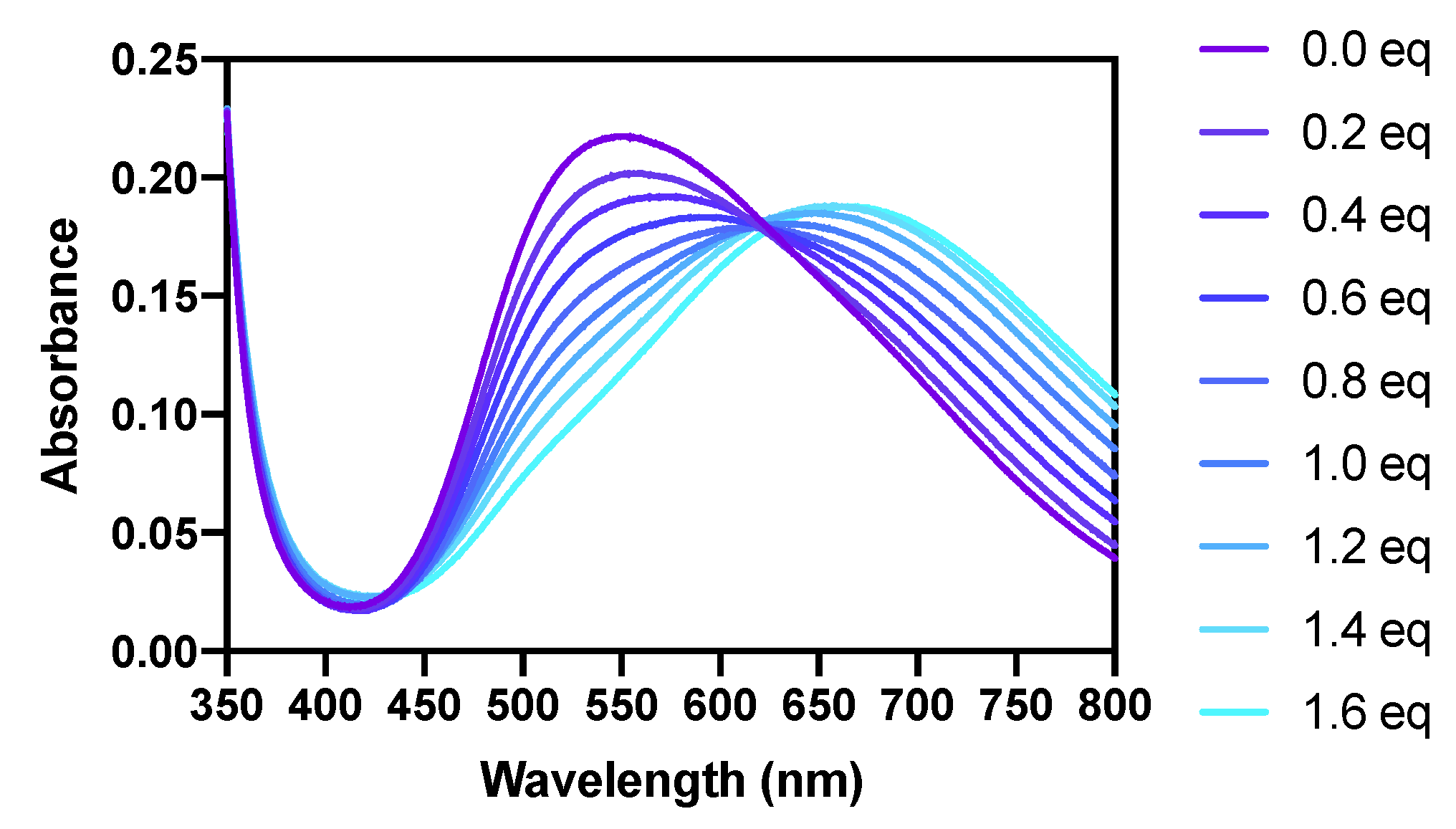
2.2. pKa Determination and Comparison
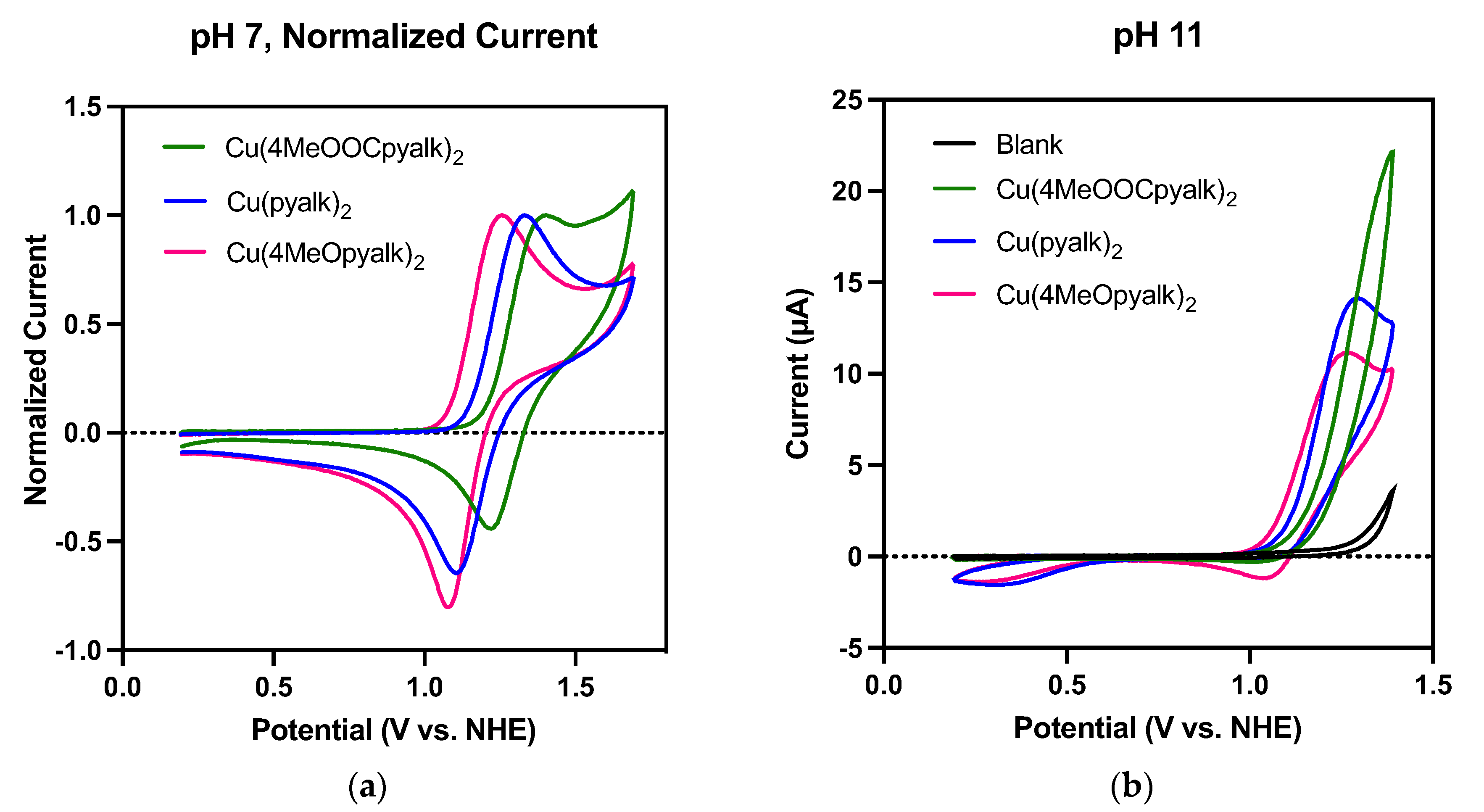
2.3. Electrochemical Studies
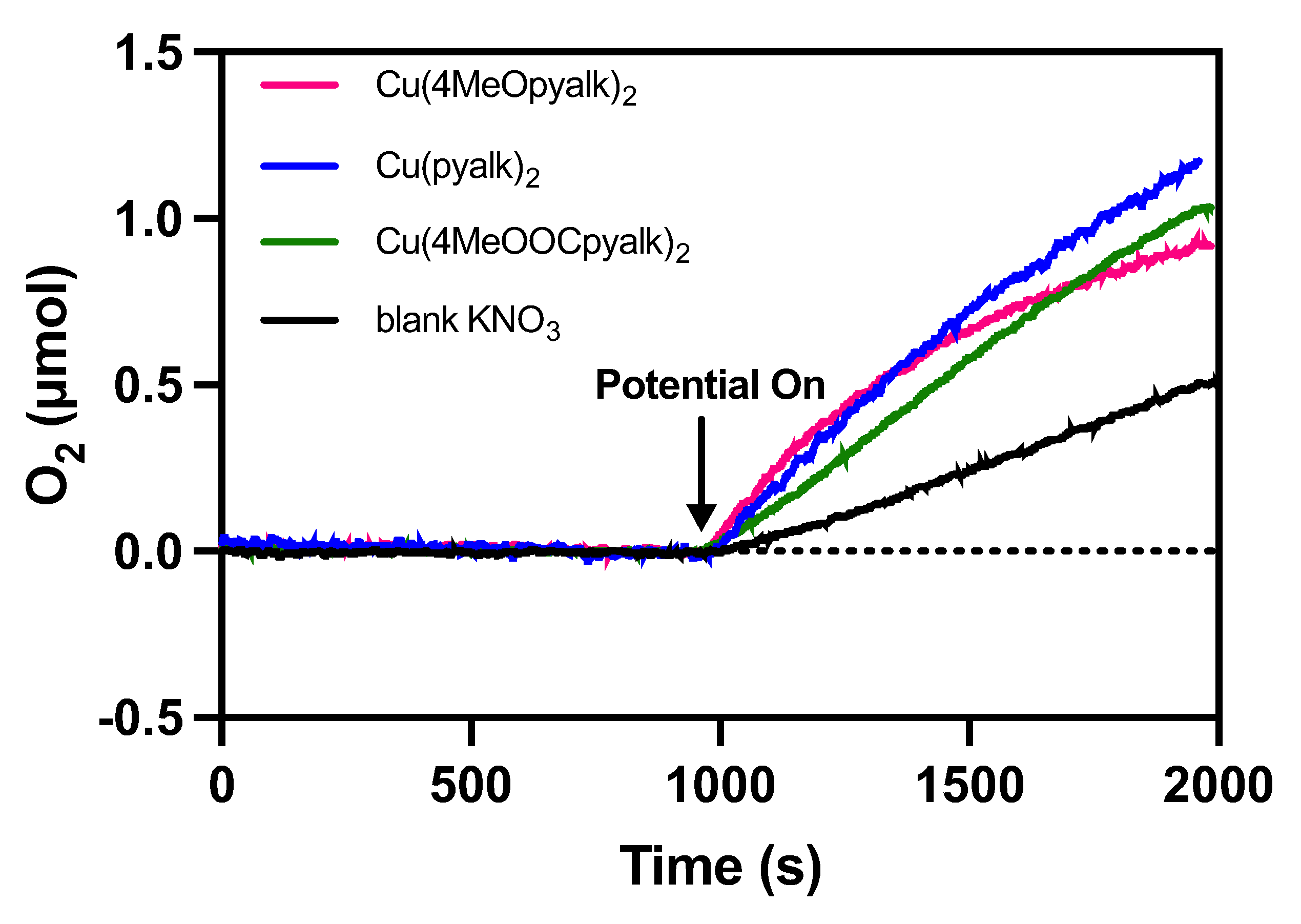
2.4. Water Oxidation Activity
3. Materials and Methods
3.1. Materials and Chemicals
3.2. Synthesis (See Scheme 1)
3.3. Instrumentation
3.4. Electrochemical Measurements
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Lewis, N.S.; Nocera, D.G. Powering the Planet: Chemical Challenges in Solar Energy Utilization. Proc. Natl. Acad. Sci. USA 2006, 103, 15729–15735. [Google Scholar] [CrossRef]
- Zhang, B.; Sun, L. Artificial Photosynthesis: Opportunities and Challenges of Molecular Catalysts. Chem. Soc. Rev. 2019, 48, 2216–2264. [Google Scholar] [CrossRef]
- Blakemore, J.D.; Crabtree, R.H.; Brudvig, G.W. Molecular Catalysts for Water Oxidation. Chem. Rev. 2015, 115, 12974–13005. [Google Scholar] [CrossRef]
- Barnett, S.M.; Goldberg, K.I.; Mayer, J.M. A Soluble Copper–Bipyridine Water-Oxidation Electrocatalyst. Nat. Chem. 2012, 4, 498–502. [Google Scholar] [CrossRef]
- Lee, H.; Wu, X.; Sun, L. Copper-Based Homogeneous and Heterogeneous Catalysts for Electrochemical Water Oxidation. Nanoscale 2020, 12, 4187–4218. [Google Scholar] [CrossRef]
- Chen, Q.-F.; Cheng, Z.-Y.; Liao, R.-Z.; Zhang, M.-T. Bioinspired Trinuclear Copper Catalyst for Water Oxidation with a Turnover Frequency up to 20000 s−1. J. Am. Chem. Soc. 2021, 143, 19761–19768. [Google Scholar] [CrossRef]
- Yu, K.; Sun, Y.; Zhu, D.; Xu, Z.; Wang, J.; Shen, J.; Zhang, Q.; Zhao, W. A Low-Cost Commercial Cu(ii)–EDTA Complex for Electrocatalytic Water Oxidation in Neutral Aqueous Solution. Chem. Commun. 2022, 58, 12835–12838. [Google Scholar] [CrossRef]
- Wu, X.; Li, F.; Zhang, B.; Sun, L. Molecular Complexes in Water Oxidation: Pre-Catalysts or Real Catalysts. J. Photochem. Photobiol. C Photochem. Rev. 2015, 25, 71–89. [Google Scholar] [CrossRef]
- Gil-Sepulcre, M.; Llobet, A. Molecular Water Oxidation Catalysts Based on First-Row Transition Metal Complexes. Nat. Catal. 2022, 5, 79–82. [Google Scholar] [CrossRef]
- Wasylenko, D.J.; Ganesamoorthy, C.; Koivisto, B.D.; Henderson, M.A.; Berlinguette, C.P. Insight into Water Oxidation by Mononuclear Polypyridyl Ru Catalysts. Inorg. Chem. 2010, 49, 2202–2209. [Google Scholar] [CrossRef]
- Maji, S.; López, I.; Bozoglian, F.; Benet-Buchholz, J.; Llobet, A. Mononuclear Ruthenium–Water Oxidation Catalysts: Discerning between Electronic and Hydrogen-Bonding Effects. Inorg. Chem. 2013, 52, 3591–3593. [Google Scholar] [CrossRef]
- Abdel-Magied, A.F.; Arafa, W.A.A.; Laine, T.M.; Shatskiy, A.; Kärkäs, M.D.; Åkermark, B.; Johnston, E.V. Substituent Effects in Molecular Ruthenium Water Oxidation Catalysts Based on Amide Ligands. Chemcatchem 2017, 9, 1583–1587. [Google Scholar] [CrossRef]
- Watabe, S.; Tanahashi, Y.; Hirahara, M.; Yamazaki, H.; Takahashi, K.; Mohamed, E.A.; Tsubonouchi, Y.; Zahran, Z.N.; Saito, K.; Yui, T.; et al. Critical Hammett Electron-Donating Ability of Substituent Groups for Efficient Water Oxidation Catalysis by Mononuclear Ruthenium Aquo Complexes. Inorg. Chem. 2019, 58, 12716–12723. [Google Scholar] [CrossRef]
- Rodriguez, G.M.; Zaccaria, F.; Van Dijk, S.; Zuccaccia, C.; Macchioni, A. Substituent Effects on the Activity of Cp*Ir(Pyridine-Carboxylate) Water Oxidation Catalysts: Which Ligand Fragments Remain Coordinated to the Active Ir Centers? Organometallics 2021, 40, 3445–3453. [Google Scholar] [CrossRef]
- Codolà, Z.; Garcia-Bosch, I.; Acuña-Parés, F.; Prat, I.; Luis, J.M.; Costas, M.; Lloret-Fillol, J. Electronic Effects on Single-Site Iron Catalysts for Water Oxidation. Chem. A Eur. J. 2013, 19, 8042–8047. [Google Scholar] [CrossRef]
- Rohner, S.S.; Kinzel, N.W.; Werlé, C.; Leitner, W. Systematic Ligand Variation to Modulate the Electrochemical Properties of Iron and Manganese Complexes. Dalton Trans. 2019, 48, 13205–13211. [Google Scholar] [CrossRef]
- Gerlach, D.L.; Bhagan, S.; Cruce, A.A.; Burks, D.B.; Nieto, I.; Truong, H.T.; Kelley, S.P.; Herbst-Gervasoni, C.J.; Jernigan, K.L.; Bowman, M.K.; et al. Studies of the Pathways Open to Copper Water Oxidation Catalysts Containing Proximal Hydroxy Groups during Basic Electrocatalysis. Inorg. Chem. 2014, 53, 12689–12698. [Google Scholar] [CrossRef]
- Garrido-Barros, P.; Funes-Ardoiz, I.; Drouet, S.; Benet-Buchholz, J.; Maseras, F.; Llobet, A. Redox Non-Innocent Ligand Controls Water Oxidation Overpotential in a New Family of Mononuclear Cu-Based Efficient Catalysts. J. Am. Chem. Soc. 2015, 137, 6758–6761. [Google Scholar] [CrossRef]
- Shen, J.; Zhang, X.; Cheng, M.; Jiang, J.; Wang, M. Electrochemical Water Oxidation Catalyzed by N 4 -Coordinate Copper Complexes with Different Backbones: Insight into the Structure-Activity Relationship of Copper Catalysts. Chemcatchem 2020, 12, 1302–1306. [Google Scholar] [CrossRef]
- Shen, J.; Wang, M.; Gao, J.; Han, H.; Liu, H.; Sun, L. Improvement of Electrochemical Water Oxidation by Fine-Tuning the Structure of Tetradentate N4 Ligands of Molecular Copper Catalysts. Chemsuschem 2017, 10, 4581–4588. [Google Scholar] [CrossRef]
- Garrido-Barros, P.; Moonshiram, D.; Gil-Sepulcre, M.; Pelosin, P.; Gimbert-Suriñach, C.; Benet-Buchholz, J.; Llobet, A. Redox Metal–Ligand Cooperativity Enables Robust and Efficient Water Oxidation Catalysis at Neutral pH with Macrocyclic Copper Complexes. J. Am. Chem. Soc. 2020, 142, 17434–17446. [Google Scholar] [CrossRef]
- de Aguirre, A.; Garrido-Barros, P.; Funes-Ardoiz, I.; Maseras, F. The Role of Electron-Donor Substituents in the Family of OPBAN-Cu Water Oxidation Catalysts: Effect on the Degradation Pathways and Efficiency. Eur. J. Inorg. Chem. 2019, 2019, 2109–2114. [Google Scholar] [CrossRef]
- Chattopadhyay, S.; Ghatak, A.; Ro, Y.; Guillot, R.; Halime, Z.; Aukauloo, A.; Dey, A. Ligand Radical Mediated Water Oxidation by a Family of Copper o-Phenylene Bis-Oxamidate Complexes. Inorg. Chem. 2021, 60, 9442–9455. [Google Scholar] [CrossRef]
- Fisher, K.J.; Materna, K.L.; Mercado, B.Q.; Crabtree, R.H.; Brudvig, G.W. Electrocatalytic Water Oxidation by a Copper(II) Complex of an Oxidation-Resistant Ligand. ACS Catal. 2017, 7, 3384–3387. [Google Scholar] [CrossRef]
- Michaelos, T.K.; Shopov, D.Y.; Sinha, S.B.; Sharninghausen, L.S.; Fisher, K.J.; Lant, H.M.C.; Crabtree, R.H.; Brudvig, G.W. A Pyridine Alkoxide Chelate Ligand That Promotes both Unusually High Oxidation States and Water-Oxidation Catalysis. Acc. Chem. Res. 2017, 50, 952–959. [Google Scholar] [CrossRef]
- Shopov, D.Y.; Sharninghausen, L.S.; Sinha, S.B.; Mercado, B.Q.; Balcells, D.; Brudvig, G.W.; Crabtree, R.H. A Dinuclear Iridium(V,V) Oxo-Bridged Complex Characterized Using a Bulk Electrolysis Technique for Crystallizing Highly Oxidizing Compounds. Inorg. Chem. 2018, 57, 5684–5691. [Google Scholar] [CrossRef]
- Fisher, K.J.; Feuer, M.L.; Lant, H.M.C.; Mercado, B.Q.; Crabtree, R.H.; Brudvig, G.W. Concerted Proton-Electron Transfer Oxidation of Phenols and Hydrocarbons by a High-Valent Nickel Complex. Chem. Sci. 2020, 11, 1683–1690. [Google Scholar] [CrossRef]
- Materna, K.L.; Rudshteyn, B.; Brennan, B.J.; Kane, M.H.; Bloomfield, A.J.; Huang, D.L.; Shopov, D.Y.; Batista, V.S.; Crabtree, R.H.; Brudvig, G.W. Heterogenized Iridium Water-Oxidation Catalyst from a Silatrane Precursor. ACS Catal. 2016, 6, 5371–5377. [Google Scholar] [CrossRef]
- Wong, Y.-L.; Yang, Q.; Zhou, Z.-Y.; Lee, H.K.; Mak, T.C.W.; Ng, D.K.P. Synthesis, Structure and Oxo-Transfer Properties of Dioxotungsten(VI) Complexes with Pyridine-Based NO-and NS-Bidentate Ligands. New J. Chem. 2001, 25, 353–357. [Google Scholar] [CrossRef]
- Rudshteyn, B.; Fisher, K.J.; Lant, H.M.C.; Yang, K.R.; Mercado, B.Q.; Brudvig, G.W.; Crabtree, R.H.; Batista, V.S. Water-Nucleophilic Attack Mechanism for the CuII(Pyalk)2 Water-Oxidation Catalyst. ACS Catal. 2018, 8, 7952–7960. [Google Scholar] [CrossRef]
- Hansch, C.; Leo, A.; Taft, R.W. A Survey of Hammett Substituent Constants and Resonance and Field Parameters. Chem. Rev. 1991, 91, 165–195. [Google Scholar] [CrossRef]
- Rountree, E.S.; McCarthy, B.D.; Eisenhart, T.T.; Dempsey, J.L. Evaluation of Homogeneous Electrocatalysts by Cyclic Voltammetry. Inorg. Chem. 2014, 53, 9983–10002. [Google Scholar] [CrossRef] [PubMed]
- Costentin, C.; Drouet, S.; Robert, M.; Savéant, J.-M. Turnover Numbers, Turnover Frequencies, and Overpotential in Molecular Catalysis of Electrochemical Reactions. Cyclic Voltammetry and Preparative-Scale Electrolysis. J. Am. Chem. Soc. 2012, 134, 11235–11242. [Google Scholar] [CrossRef]
- Shopov, D.Y.; Sharninghausen, L.S.; Sinha, S.B.; Mercado, B.Q.; Brudvig, G.W.; Crabtree, R.H. Modification of a Pyridine-Alkoxide Ligand during the Synthesis of Coordination Compounds. Inorganica Chim. Acta 2019, 484, 75–78. [Google Scholar] [CrossRef]
- Sheldrick, G.M. A Short History of SHELX. Acta Crystallogr. Sect. A Found. Crystallogr. 2008, 64, 112–122. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 1 1 | 2 | 3 | |
|---|---|---|---|
| Cu–N1 (Å) | 1.9724 (13) | 1.9696 (17) | 1.945 (13) |
| Cu–O1 (Å) | 1.8833 (11) | 1.8777 (14) | 1.8866 (11) |
| O1–Cu–N1 (°) | 84.04 (5) | 84.29 (7) | 84.33 (5) |
| Complex | pKa | λmax (nm) |
|---|---|---|
| Cu(4-MeOpyalk)2 (3) | 8.2 ± 0.1 | 554 |
| Cu(pyalk)2 (1) | 7.91 ± 0.08 | 547 |
| Cu(4-MeOOCpyalk)2 (2) | 7.57 ± 0.08 | 543 |
| Complex | E1/2 (V vs. NHE) | Ep/2 (V vs. NHE) | η (mV) |
|---|---|---|---|
| Cu(4-MeOpyalk)2 (3) | 1.17 | 1.13 | 550 |
| Cu(pyalk)2 (1) | 1.21 | 1.18 | 600 |
| Cu(4-MeOOCpyalk)2 (2) | 1.31 | 1.26 | 680 |
| Complex | Initial O2 Rate (μmol s−1) | Faradaic Efficiency (%) |
|---|---|---|
| Cu(4-MeOpyalk)2 (3) | 0.0020 | 71 |
| Cu(pyalk)2 (1) | 0.0019 | 90 |
| Cu(4-MeOOCpyalk)2 (2) | 0.0011 | 79 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cody, C.C.; Caes, Z.N.; Capobianco, M.D.; Mercado, B.Q.; Crabtree, R.H.; Brudvig, G.W. Ligand Tuning in Cu(pyalk)2 Water Oxidation Electrocatalysis. Inorganics 2023, 11, 229. https://doi.org/10.3390/inorganics11060229
Cody CC, Caes ZN, Capobianco MD, Mercado BQ, Crabtree RH, Brudvig GW. Ligand Tuning in Cu(pyalk)2 Water Oxidation Electrocatalysis. Inorganics. 2023; 11(6):229. https://doi.org/10.3390/inorganics11060229
Chicago/Turabian StyleCody, Claire C., Zofia N. Caes, Matt D. Capobianco, Brandon Q. Mercado, Robert H. Crabtree, and Gary W. Brudvig. 2023. "Ligand Tuning in Cu(pyalk)2 Water Oxidation Electrocatalysis" Inorganics 11, no. 6: 229. https://doi.org/10.3390/inorganics11060229