
Chalcogen Bonds, Halogen Bonds and Halogen···Halogen Contacts in Di- and Tri-iododiorganyltellurium(IV) Derivatives


Abstract
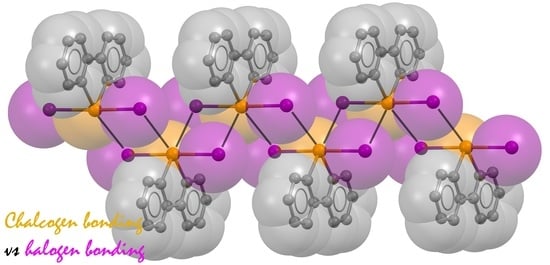
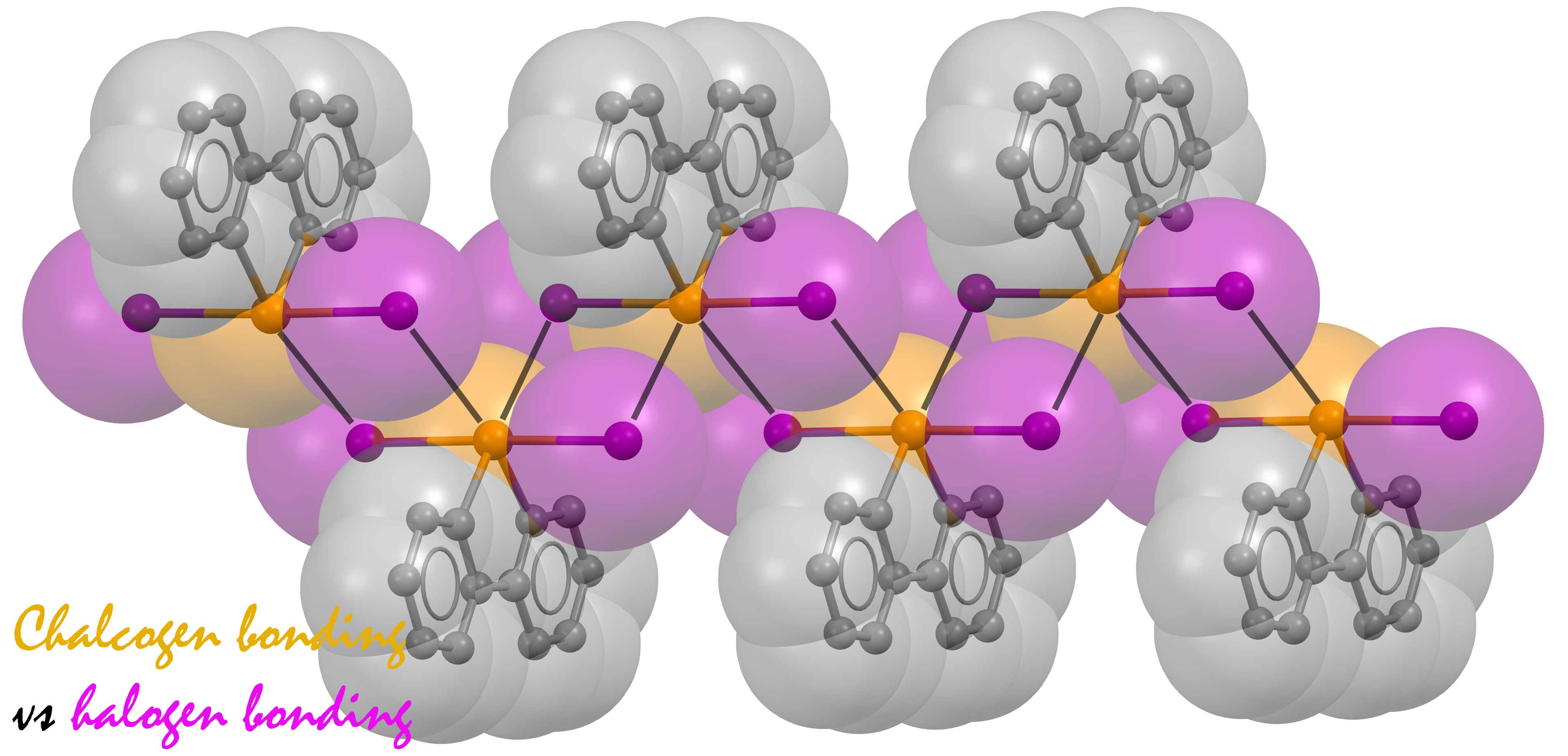
:
1. Introduction
2. Results and Discussion
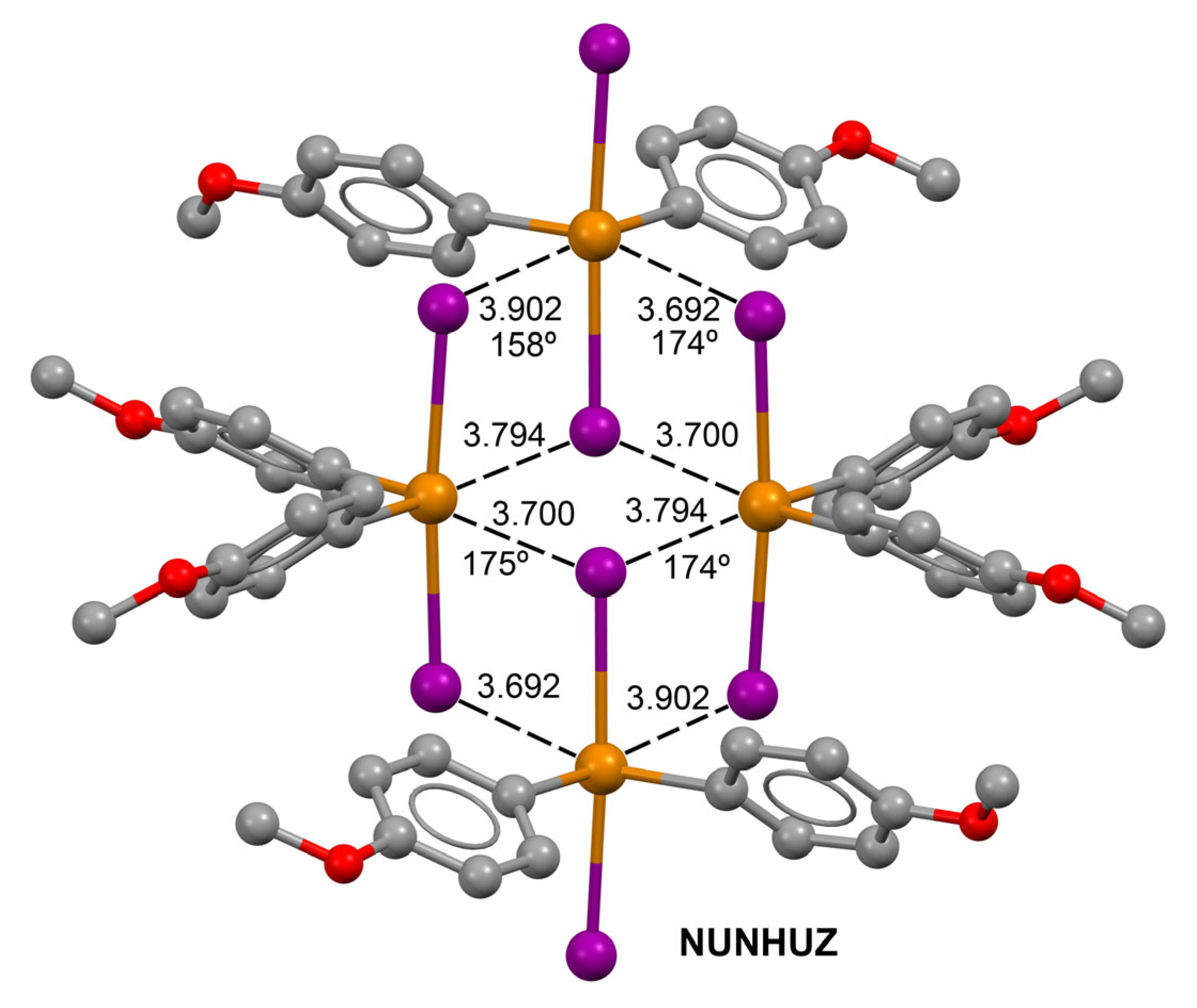
2.1. CSD Survey
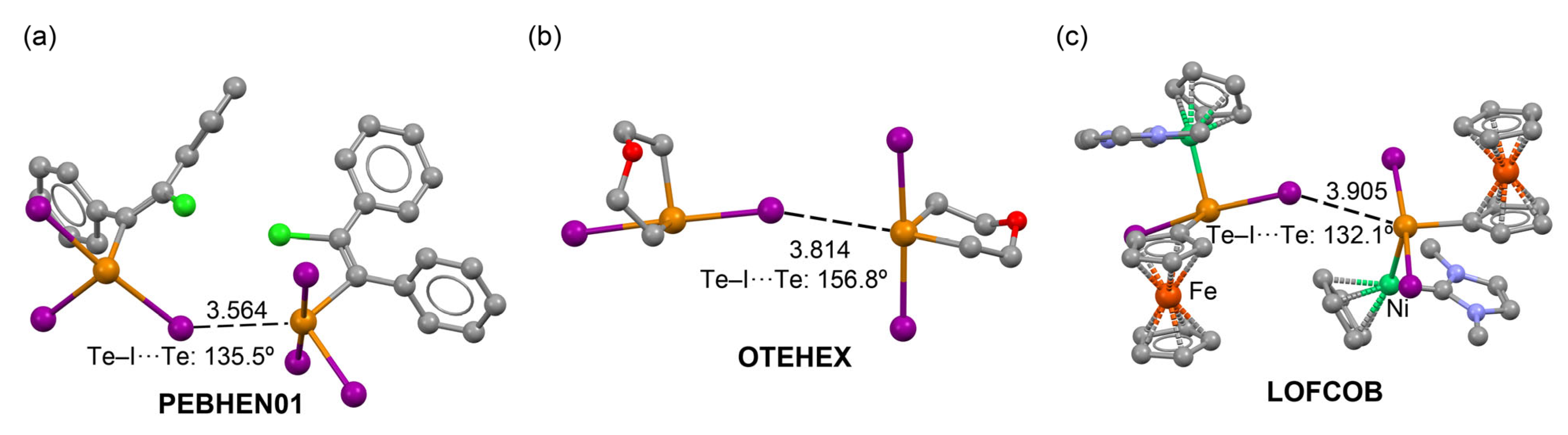
ChB Assemblies
2.2. Theoretical Study
2.2.1. C–Te···I ChBs
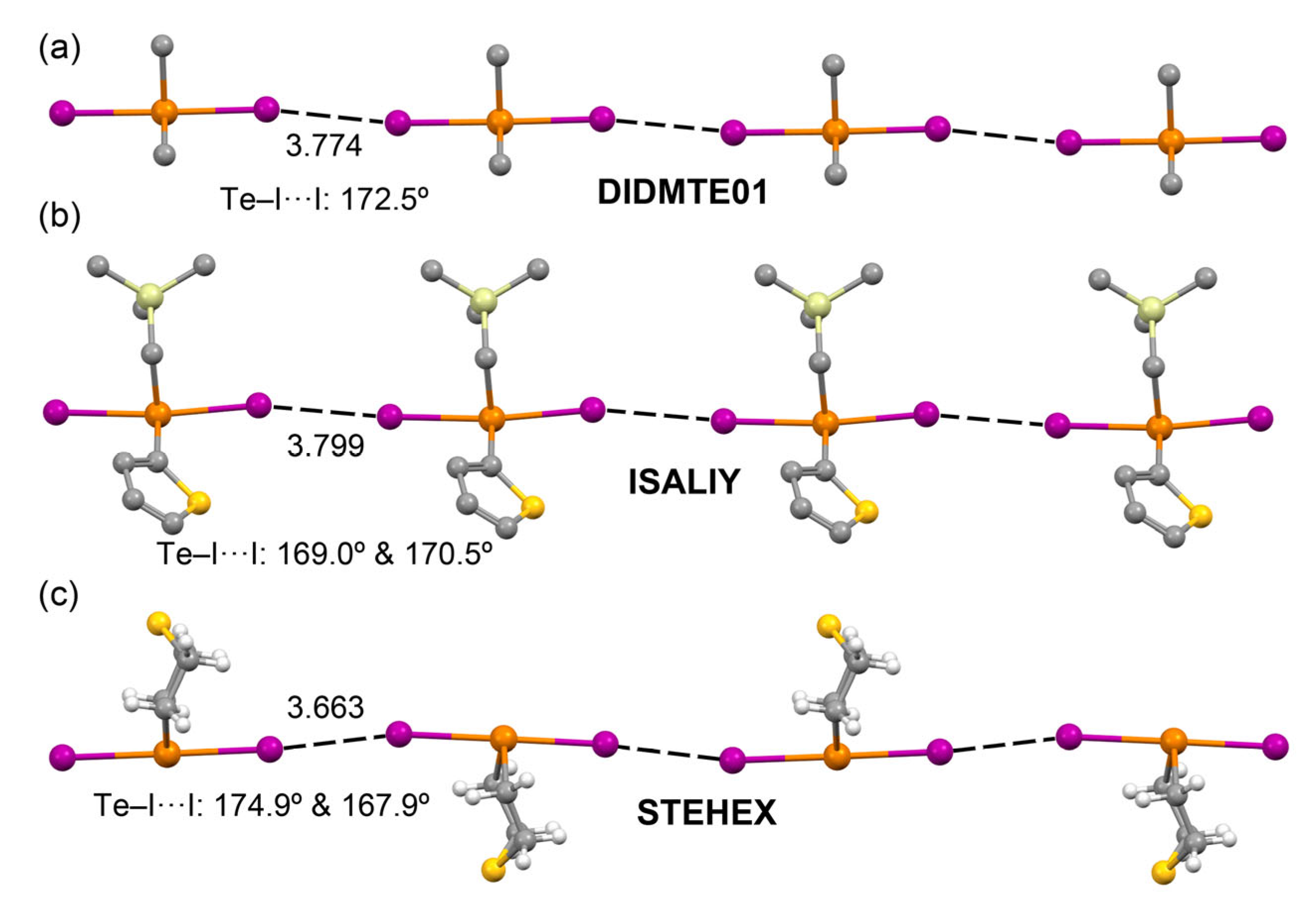
2.2.2. Te–I···I–Te Type I Halogen···Halogen Interactions
2.2.3. Te–I···I–Te Halogen-Bonding Interactions
2.2.4. Uncategorized Te···I Contacts
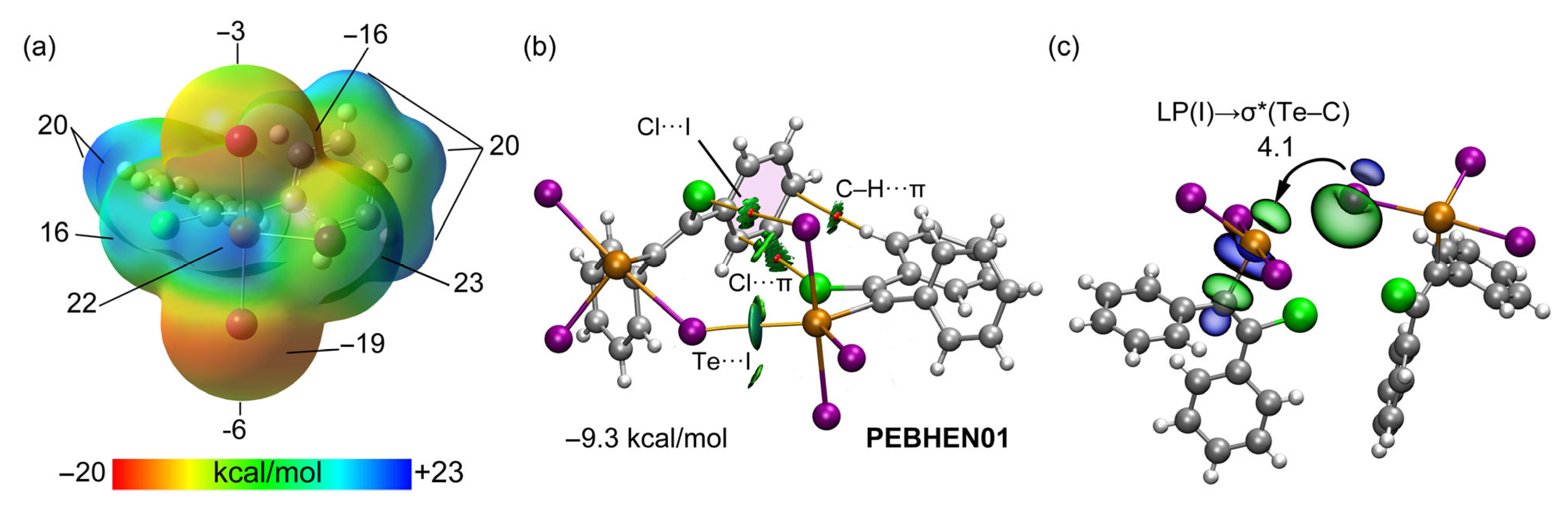
2.2.5. Energy Decomposition Analysis
3. Computational Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Desiraju, G.R.; Ho, P.S.; Kloo, L.; Legon, A.C.; Marquardt, R.; Metrangolo, P.; Politzer, P.; Resnati, G.; Rissanen, K. Definition of the halogen bond (IUPAC Recommendations 2013). Pure Appl. Chem. 2013, 85, 1711–1713. [Google Scholar] [CrossRef]
- Aakeroy, C.B.; Bryce, D.L.; Desiraju, G.R.; Frontera, A.; Legon, A.C.; Nicotra, F.; Rissanen, K.; Scheiner, S.; Terraneo, G.; Metrangolo, P.; et al. Definition of the chalcogen bond (IUPAC Recommendations 2019). Pure Appl. Chem. 2019, 91, 1889–1892. [Google Scholar] [CrossRef]
- Zahn, S.; Frank, R.; Hey-Hawkins, E.; Kirchner, B. Pnicogen bonds: A new molecular linker? Chem. Eur. J. 2011, 17, 6034–6038. [Google Scholar] [CrossRef]
- Bauzá, A.; Mooibroek, T.J.; Frontera, A. Tetrel Bonding Interaction: Rediscovered Supramolecular Force? Angew. Chem. Int. Ed. 2013, 52, 12317–12321. [Google Scholar] [CrossRef] [PubMed]
- Bauzá, A.; Frontera, A. σ/π-Hole noble gas bonding interactions: Insights from theory and experiment. Coord. Chem. Rev. 2020, 404, 213112. [Google Scholar] [CrossRef]
- Bauzá, A.; Frontera, A.; Mooibroek, T.J. NO3− anions can act as Lewis acid in the solid state. Nat. Commun. 2017, 8, 14522. [Google Scholar] [CrossRef]
- García-Llinás, X.; Bauzá, A.; Seth, S.K.; Frontera, A. Importance of R–CF3···O Tetrel Bonding Interactions in Biological Systems. J. Phys. Chem. A 2017, 28, 5371–5376. [Google Scholar] [CrossRef]
- Bauzá, A.; Sharko, A.V.; Senchyk, G.A.; Rusanov, E.B.; Frontera, A.; Domasevitch, K.V. π–hole interactions at work: Crystal engineering with nitro-derivatives. Cryst. Eng. Comm. 2017, 19, 1933–1937. [Google Scholar] [CrossRef]
- Bauzá, A.; Frontera, A.; Mooibroek, T.J. π-hole interactions involving nitro compounds: Directionality of nitrate esters. Cryst. Growth Des. 2016, 16, 5520–5524. [Google Scholar] [CrossRef]
- Politzer, P.; Murray, J. σ-Hole Interactions: Perspectives and Misconceptions. Crystals 2017, 7, 212. [Google Scholar] [CrossRef]
- Politzer, P.; Murray, J.S.; Clark, T. Halogen bonding and other σ-hole interactions: A perspective. Phys. Chem. Chem. Phys. 2013, 15, 11178–11189. [Google Scholar] [CrossRef] [PubMed]
- Bauza, A.; Mooibroek, T.J.; Frontera, A. The Bright Future of Unconventional σ/π-Hole Interactions. Chem. Phys. Chem. 2015, 16, 2496–2517. [Google Scholar] [CrossRef]
- Riwar, L.J.; Trapp, N.; Root, K.; Zenobi, R.; Diederich, F. Supramolecular Capsules: Strong versus Weak Chalcogen Bonding. Angew. Chem. Int. Ed. 2018, 57, 17259–17264. [Google Scholar] [CrossRef] [PubMed]
- Wonner, P.; Dreger, A.; Vogel, L.; Engelage, E.; Huber, S.M. Chalcogen Bonding Catalysis of a Nitro-Michael Reaction. Angew. Chem. Int. Ed. 2019, 58, 16923–16927. [Google Scholar] [CrossRef]
- Borissov, A.; Marques, I.; Lim, J.Y.C.; Félix, V.; Smith, M.D.; Beer, P.D. Anion Recognition in Water by Charge-Neutral Halogen and Chalcogen Bonding Foldamer Receptors. J. Am. Chem. Soc. 2019, 141, 4119–4129. [Google Scholar] [CrossRef]
- Mallada, B.; Gallardo, A.; Lamanec, M.; De La Torre, B.; Špirko, V.; Hobza, P.; Jelinek, P. Real-space imaging of anisotropic charge of σ-hole by means of Kelvin probe force microscopy. Science 2021, 374, 863–867. [Google Scholar] [CrossRef]
- Pascoe, J.; Ling, K.B.; Cockroft, S.L. The Origin of Chalcogen-Bonding Interactions. J. Am. Chem. Soc. 2017, 139, 15160–15167. [Google Scholar] [CrossRef]
- Macchione, M.; Goujon, A.; Strakova, K.; Humeniuk, H.V.; Licari, G.; Tajkhorshid, E.; Sakai, N.; Matile, S. A Chalcogen-Bonding Cascade Switch for Planarizable Push-Pull Probes. Angew. Chem. Int. Ed. 2019, 58, 15752–15756. [Google Scholar] [CrossRef]
- Politzer, P.; Murray, J.S. An overview of strengths and directionalities of noncovalent interactions: σ-holes and π-holes. Crystals 2019, 9, 165. [Google Scholar] [CrossRef]
- Politzer, P.; Murray, J.S.; Clark, T.; Resnati, G. The σ-hole revisited. Phys. Chem. Chem. Phys. 2017, 19, 32166–32178. [Google Scholar] [CrossRef] [PubMed]
- Murray, J.S.; Lane, P.; Clark, T.; Riley, K.E.; Politzer, P. σ-Holes, π-holes and electrostatically-driven interactions. J. Mol. Mod. 2012, 18, 541–548. [Google Scholar] [CrossRef] [PubMed]
- Cavallo, G.; Metrangolo, P.; Pilati, T.; Resnati, G.; Terraneo, G. Naming interactions from the electrophilic site. Cryst. Growth Des. 2014, 14, 2697–2702. [Google Scholar] [CrossRef]
- Terraneo, G.; Resnati, G. Bonding Matters. Cryst. Growth Des. 2017, 17, 1439–1440. [Google Scholar] [CrossRef]
- Scheiner, S. Steric crowding in tetrel bonds. J. Phys. Chem. A 2018, 122, 2550–2562. [Google Scholar] [CrossRef]
- Bauzá, A.; Mooibroek, T.J.; Frontera, A. Tetrel Bonding Interactions. Chem. Rec. 2016, 16, 473–487. [Google Scholar] [CrossRef] [PubMed]
- Mani, D.; Arunan, E. The X–C⋯Y (X = O/F, Y = O/S/F/Cl/Br/N/P) ‘carbon bond’ and hydrophobic interactions. Phys. Chem. Chem. Phys. 2013, 15, 14377–14383. [Google Scholar] [CrossRef]
- Sethio, D.; Oliveira, V.; Kraka, E. Quantitative Assessment of Tetrel Bonding Utilizing Vibrational Spectroscopy. Molecules 2018, 23, 2763. [Google Scholar] [CrossRef]
- Heywood, V.L.; Alford, T.P.J.; Roeleveld, J.J.; Deprez, S.J.L.; Verhoofstad, A.; van der Vlugt, J.I.; Domingos, S.R.; Schnell, M.; Davis, A.P.; Mooibroek, T.J. Observations of tetrel bonding between sp3-carbon and THF. Chem. Sci. 2020, 11, 5289–5293. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, W.; Wang, Y.-B. Tetrel bonding on Graphene. Comp. Theor. Chem. 2019, 1147, 8–12. [Google Scholar] [CrossRef]
- Taylor, M.S. Anion recognition based on halogen, chalcogen, pnictogen and tetrel bonding. Coord. Chem. Rev. 2020, 413, 213720. [Google Scholar] [CrossRef]
- Bauzá, A.; Seth, S.K.; Frontera, A. Tetrel bonding interactions at work: Impact on tin and lead coordination compounds. Coord. Chem. Rev. 2019, 384, 107–125. [Google Scholar] [CrossRef]
- Mundlapati, V.R.; Sahoo, D.K.; Bhaumik, S.; Jena, S.; Chandrakar, A.; Biswal, H.S. Noncovalent Carbon-Bonding Interactions in Proteins. Angew. Chem. Int. Ed. 2018, 57, 16496. [Google Scholar] [CrossRef] [PubMed]
- Scilabra, P.; Terraneo, G.; Resnati, G. The Chalcogen Bond in Crystalline Solids: A World Parallel to Halogen Bond. Acc. Chem. Res. 2019, 52, 1313–1324. [Google Scholar] [CrossRef] [PubMed]
- Fourmigué, M.; Dhaka, A. Chalcogen bonding in crystalline diselenides and selenocyanates: From molecules of pharmaceutical interest to conducting materials. Coord. Chem. Rev. 2020, 403, 213084. [Google Scholar] [CrossRef]
- Alkorta, I.; Elguero, J.; Frontera, A. Not only hydrogen bonds: Other noncovalent interactions. Crystals 2020, 10, 180. [Google Scholar] [CrossRef]
- Vogel, L.; Wonner, P.; Huber, S.M. Chalcogen bonding: An overview. Angew. Chem. Int. Ed. 2019, 58, 1880–1891. [Google Scholar] [CrossRef] [PubMed]
- Biot, N.; Bonifazi, D. Chalcogen-bond driven molecular recognition at work. Coord. Chem. Rev. 2020, 413, 213243. [Google Scholar] [CrossRef]
- Navarro-García, E.; Galmés, B.; Velasco, M.D.; Frontera, A.; Caballero, A. Anion Recognition by Neutral Chalcogen Bonding Receptors: Experimental and Theoretical Investigations. Chem. Eur. J. 2020, 26, 4706–4713. [Google Scholar] [CrossRef] [PubMed]
- Navarro-García, E.; Galmés, B.; Velasco, M.D.; Bastida, A.; Zapata, F.; Caballero, A.; Frontera, A. Host–guest complexes vs. supramolecular polymers in chalcogen bonding receptors: An experimental and theoretical study. Dalton Trans. 2022, 51, 1325–1332. [Google Scholar] [CrossRef]
- Frontera, A.; Bauza, A. Metal Coordination Enhances Chalcogen Bonds: CSD Survey and Theoretical Calculations. Int. J. Mol. Sci. 2022, 23, 4188. [Google Scholar] [CrossRef]
- Mahmudov, K.T.; Kopylovich, M.N.; Guedes da Silva, M.F.C.; Pombeiro, A.J.L. Chalcogen bonding in synthesis, catalysis and design of materials. Dalton Trans. 2017, 46, 10121–10138. [Google Scholar] [CrossRef] [PubMed]
- Romito, D.; Fresta, E.; Cavinato, L.M.; Kählig, H.; Amenitsch, H.; Caputo, L.; Chen, Y.; Samorì, P.; Charlier, J.-C.; Costa, R.D.; et al. Supramolecular Chalcogen-Bonded Semiconducting Nanoribbons at Work in Lighting Devices. Angew. Chem. Int. Ed. 2022, 61, e202202137. [Google Scholar] [CrossRef] [PubMed]
- Adhikari, U.; Scheiner, S. Effects of Charge and Substituent on the S···N Chalcogen Bond. J. Phys. Chem. A 2014, 118, 3183–3192. [Google Scholar] [CrossRef]
- Goodman, M.A.; Detty, M.R. Selenoxides as Catalysts for the Activation of Hydrogen Peroxide. Bromination of Organic Substrates with Sodium Bromide and Hydrogen Peroxide. Organometallics 2004, 23, 3016–3020. [Google Scholar] [CrossRef]
- Farina, M.; Folmer, V.; Bolzan, R.C.; Andrade, L.H.; Zeni, G.; Braga, A.J.; Rocha, J.B.T. Selenoxides inhibit δ-aminolevulinic acid dehydratase. Toxicol. Lett. 2001, 119, 27–37. [Google Scholar] [CrossRef]
- Franconetti, A.; Quiñonero, D.; Frontera, A.; Resnati, G. Unexpected chalcogen bonds in tetravalent sulfur compounds. Phys. Chem. Chem. Phys. 2019, 21, 11313–11319. [Google Scholar] [CrossRef] [PubMed]
- Alcock, N.W.; Harrison, W.D. Secondary bonding. Part 12. Aryltellurium iodides: Crystal and molecular structures of cis- and trans-phenyltellurium(IV) tri-iodide and two modifications of diphenyltellurium(IV) di-iodide. J. Chem. Soc. Dalton Trans. 1984, 869–875. [Google Scholar] [CrossRef]
- Raatikainen, K.; Rissanen, K. Hierarchical halogen bonding induces polymorphism. CrystEngComm 2009, 11, 750–752. [Google Scholar] [CrossRef]
- Haddad, S.F.; Willett, R.D.; Twamley, B. The Role of Hydrogen Bonding and Halogen Bonding in the Polymorphic Structures of 3,5-Dibromo-2,6-diaminopyridinium Bromide. J. Chem. Crystallogr. 2010, 40, 902–908. [Google Scholar] [CrossRef]
- Torubaev, Y.V.; Skabitsky, I.V. Crystals at a Carrefour on the Way through the Phase Space: A Middle Path. Molecules 2021, 26, 1583. [Google Scholar] [CrossRef]
- Hargittai, J.; Rozsondai, B. The Chemistry of Organic Selenium and Tellurium Compounds; Patai, S., Rappoport, Z., Eds.; Wiley: New York, NY, USA, 1986; Volume 1, p. 63. [Google Scholar]
- Lrgolic, K.J. Synthetic Methods of Organometallic and Inorganic Chemistry; Herrmann, W.A., Cybill, C.E., Eds.; Georg Thieme Verlag: New York, NY, USA, 1997; Volume 4, p. 219. [Google Scholar]
- Närhi, S.M.; Oilunkaniemi, R.; Laitinen, R.S.; Ahlgrén, M. The Reactions of Tellurium Tetrahalides with Triphenylphosphine under Ambient Conditions. Inorg. Chem. 2004, 43, 3742–3750. [Google Scholar] [CrossRef] [PubMed]
- Chauhan, A.K.S.; Kumar, A.; Srivastava, R.C.; Butcher, R.J. Synthesis and characterization of monomeric diorganotellurium dihalides: Crystal and molecular structures of diphenacyltellurium dibromide and-diiodide. J. Organomet. Chem. 2002, 658, 169–175. [Google Scholar] [CrossRef]
- Asahara, M.; Taomolo, S.; Tanaka, M.; Erabi, T.; Wada, M. Dependence of the rotational barrier of the Ar-group in RArTeX2 on the R-group [Ar = 2,6-(MeO)2C6H3; R = Me, Et, i-Pr; X = Cl, Br, I]. Dalton Trans. 2003, 973–979. [Google Scholar] [CrossRef]
- Hesford, M.J.; Levason, W.; Matthews, M.L.; Orchard, S.D.; Reid., G. Synthesis, characterisation and coordinating properties of the small ring S2Te-donor macrocycles [9]aneS2Te, [11]aneS2Te and [12]aneS2Te. Dalton Trans. 2003, 2434–2442. [Google Scholar] [CrossRef]
- Beckmann, J.; Dakternicks, O.; Duthic, A.; Mitchell, C.; Schurmann, M. Observation of Te∙∙∙π and X∙∙∙X Bonding in para-Substituted Diphenyltellurium Dihalides, (p-Me2NC6H4)(p-YC6H4)TeX2 (X = Cl, Br, I., Y = H, EtO, Me2N). Aust. J. Chem 2005, 58, 119–127. [Google Scholar] [CrossRef]
- Chauhan, A.K.S.; Annmika; Kumar, A.; Srivnstava, R.C.; Butcher, R.J. Secondary bonding in functionalized organotellurium compounds: Preparation and structural characterization of bis(acetamido)tellurium(IV) diiodide and bis(4-methylbenzoylmethyl)tellurium(II). J. Organomet. Chem. 2005, 690, 313–321. [Google Scholar] [CrossRef]
- Chauhan, A.K.S.; Anamika; Kumar, A.; Srivastava, R.C.; Butcher, R.J.; Beckmann, J.; Duthie, A. The interplay of secondary Te⋯N, Te⋯O, Te⋯I and I⋯I interactions, Te⋯π contacts and π-stacking in the supramolecular structures of [{2-(4-nitrobenzylideneamino)-5-methyl}phenyl](4-methoxyphenyl)tellurium dihalides. J. Organomet. Chem. 2005, 690, 1350–1355. [Google Scholar] [CrossRef]
- Block, E.; Dikarev, E.V.; Glass, R.S.; Jin, J.; Li, B.; Li, X.; Zhang, S.-Z. Synthesis, Structure, and Chemistry of New, Mixed Group 14 and 16 Heterocycles: Nucleophile-Induced Ring Contraction of Mesocyclic Dications. J. Am. Chem. Soc. 2006, 128, 14949–14961. [Google Scholar] [CrossRef]
- Srivastava, P.C.; Bajpai, S.; Bajpai, S.; Kumar, R.; Srivastava, S.; Butcher, R. Modification of supramolecular assemblies based on C4H7(CH3)Te heterocycle and cooperative participation of intermolecular I···I, Te···I, Te···O secondary bonds; C(sp3)–H···O and C(sp2)–H···O hydrogen bonds. J. Struct. Chem. 2007, 18, 223–230. [Google Scholar] [CrossRef]
- Chauhan, A.K.S.; Singh, P.; Kumar, A.; Srivastava, R.C.; Butcher, R.J.; Duthic, A. Room-Temperature Insertion of Elemental Tellurium into the Csp3−Br and −I Bonds of α-Bromo- and α-Iodopinacolone. Organometallics 2007, 26, 1955–1959. [Google Scholar] [CrossRef]
- Chauhan, A.K.S.; Singh, P.; Srivastavn, R.C.; Duthie, A.; Voda, A. Acylmethyl(aryl)tellurium(IV,II) derivatives: Intramolecular secondary bonding and steric rigidity. Dalton Trans 2008, 4023–4028. [Google Scholar] [CrossRef] [PubMed]
- Gurnani, C.; Levason, W.; Ratnani, R.; Reid, C.; Webster, M. Synthesis, characterisation and structures of thio-, seleno-and telluro-ether complexes of gallium (III). Dalton Trans. 2008, 6274–6282. [Google Scholar] [CrossRef] [PubMed]
- Groom, C.R.; Bruno, I.J.; Lightfoot, M.P.; Ward, S.C. The Cambridge Structural Database. Acta Cryst. 2016, B72, 171–179. [Google Scholar] [CrossRef] [PubMed]
- McCullough, J.D. Crystal and molecular structure of dibenzotellurophene diiodide C12H8TeI2. Inorg. Chem. 1975, 14, 1142–1146. [Google Scholar] [CrossRef]
- Srivastava, P.C.; Singh, V.; Dwivedi, S.; Butcher, R.J. Molecular and supramolecular structures of diethyltellurium diiodide and diethyltellurium bis(carboxylates). Indian J. Chem. 2010, A49, 1197–1205. [Google Scholar]
- Farran, J.; Alvarez-Larena, A.; Capparelli, M.V.; Piniella, J.F.; Germain, G. Torres-Castellanos, L. Two Polymorphs of Bis(4-methoxyphenyl)tellurium(IV) Diiodide. Acta Cryst. 1998, C54, 995–1000. [Google Scholar]
- Knight, F.R.; Fuller, A.L.; Buhl, M.; Slawin, A.M.Z.; Woollins, J.D. Hypervalent Adducts of Chalcogen-Containing peri-Substituted Naphthalenes; Reactions of Sulfur, Selenium, and Tellurium with Dihalogens. Inorg. Chem. 2010, 49, 7577–7596. [Google Scholar] [CrossRef]
- Knight, F.R.; Arachchige, K.S.A.; Randall, R.A.M.; Buhl, M.; Slawin, A.M.Z.; Woollins, J.D. Exploring hypervalency and three-centre, four-electron bonding interactions: Reactions of acenaphthenechalcogen donors and dihalogen acceptors. Dalton Trans. 2012, 41, 3154–3165. [Google Scholar] [CrossRef]
- Lang, E.S.; de Oliveira, G.M.; Silveira, E.T.; Burrow, R.A.; Vazquez-Lopez, E.M. Crystal and molecular structure of (α-naphthyl)TeI3. J. Organomet. Chem. 2002, 664, 306–309. [Google Scholar] [CrossRef]
- Desiraju, G.R.; Parthasarathy, R. The nature of halogen···halogen interactions: Are short halogen contacts due to specific attractive forces or due to close packing of nonspherical atoms? J. Am. Chem. Soc. 1989, 111, 8725–8726. [Google Scholar] [CrossRef]
- Pedireddi, V.R.; Reddy, D.S.; Goud, B.S.; Craig, D.C.; Rae, A.D.; Desiraju, G.R. The nature of halogen ··· halogen interactions and the crystal structure of 1,3,5,7-tetraiodoadamantane. J. Chem. Soc. Perkin Trans. 1994, 2, 2353–2360. [Google Scholar] [CrossRef]
- Fourmigué, M. Halogen bonding: Recent advances. Curr. Opin. Solid State Mater. Sci. 2009, 13, 36–45. [Google Scholar] [CrossRef]
- Bauzá, A.; Frontera, A. Competition between lone pair-π, halogen-π and triel bonding interactions involving BX3 (X = F, Cl, Br and I) compounds: An ab initio study. Theor. Chem. Acc. 2017, 136, 37. [Google Scholar] [CrossRef]
- Cavallo, G.; Metrangolo, P.; Milani, R.; Pilati, T.; Priimagi, A.; Resnati, G.; Terraneo, G. The Halogen Bond. Chem. Rev. 2016, 116, 2478–2601. [Google Scholar] [CrossRef] [PubMed]
- Grabowski, S.J. Halogen bond with the multivalent halogen acting as the Lewis acid center. Chem. Phys. Lett 2014, 605−606, 131–136. [Google Scholar] [CrossRef]
- Adhikari, U.; Scheiner, S. Sensitivity of pnicogen, chalcogen, halogen and H-bonds to angular distortions. Chem. Phys. Lett. 2012, 532, 31–35. [Google Scholar] [CrossRef]
- Scheiner, S. Sensitivity of Noncovalent Bonds to Intermolecular Separation: Hydrogen, Halogen, Chalcogen, and Pnicogen Bonds. CrystEngComm 2013, 15, 3119–3124. [Google Scholar] [CrossRef]
- Clark, T.; Hennemann, M.; Murray, J.S.; Politzer, P. Halogen bonding: The σ-hole. J. Mol. Model. 2007, 13, 291–296. [Google Scholar] [CrossRef]
- Nziko, V.D.P.N.; Scheiner, S. Comparison of π-hole tetrel bonding with σ-hole halogen bonds in complexes of XCN (X = F, Cl,Br, I) and NH3. Phys. Chem. Chem. Phys. 2016, 18, 3581–3590. [Google Scholar] [CrossRef]
- Scheiner, S. Characterization of Type I and II Interactions between Halogen Atoms. Cryst. Growth Des. 2022, 22, 2692–2702. [Google Scholar] [CrossRef]
- Mukherjee, A.; Tothadi, S.; Desiraju, G.R. Halogen Bonds in Crystal Engineering: Like Hydrogen Bonds yet Different. Acc. Chem. Res. 2014, 47, 2514–2524. [Google Scholar] [CrossRef] [PubMed]
- Nyburg, S.C.; Faerman, C.H. A revision of van der Waals atomic radii for molecular crystals: N, O, F, S, Cl, Se, Br and I bonded to carbon. Acta Crystallogr. Sect. B Struct. Sci. 1985, 41, 274–279. [Google Scholar] [CrossRef]
- Nyburg, S.C. Intermolecular Forces and the Space Group of Solid Chlorine. J. Chem. Phys. 1964, 40, 2493–2501. [Google Scholar] [CrossRef]
- Spilfogel, T.S.; Titi, H.M.; Friščić, T. Database Investigation of Halogen Bonding and Halogen···Halogen Interactions between Porphyrins: Emergence of Robust Supramolecular Motifs and Frameworks. Cryst. Growth Des. 2021, 21, 1810–1832. [Google Scholar] [CrossRef]
- Bui, T.T.T.; Dahaoui, S.; Lecomte, C.; Desiraju, G.R.; Espinosa, E. The Nature of Halogen···Halogen Interactions: A Model Derived from Experimental Charge-Density Analysis. Angew. Chem. Int. Ed. 2009, 48, 3838–3841. [Google Scholar] [CrossRef]
- Poropudas, M.J.; Vigo, L.; Oilunkaniemi, R.; Laitinen, R.S. Structural trends in TeI2RR′: Crystal structures and NMR spectra of TeI2(CH2SiMe3)2, TeI2Th(CH2SiMe3), TeI2Ph(CH2SiMe3), and TeI2Th2 (Th = 2-Thienyl, C4H3S). Heteroat. Chem. 2011, 22, 348–359. [Google Scholar] [CrossRef]
- Knobler, C.; McCullough, J.D.; Hope, H. Crystal and molecular structure of 1-thia-4-telluracyclohexane 4,4-diiodide, C4H8STeI2. Inorg. Chem. 1970, 9, 797–804. [Google Scholar] [CrossRef]
- Torubaev, Y.V.; Samigullina, A.S. Long-Range Supramolecular Synthon Isomerism: Insight from a Case Study of Vinylic Tellurium Trihalides Cl(Ph)C=C(Ph)TeX3 (X = Cl, I). Chemistry 2022, 4, 196–205. [Google Scholar] [CrossRef]
- Hope, H.; McCullough, J.D.; Knobler, C. Crystal and molecular structure of 1-oxa-4-telluracyclohexane 4,4-diiodide, C4H8OTeI2. Inorg. Chem. 1973, 12, 2665–2669. [Google Scholar] [CrossRef]
- Shapovalov, S.S.; Pasynskii, A.A.; Skabitskii, I.V.; Tikhonova, O.G.; Kolos, A.V.; Grigor’ev, M.O. Chalcogenide Complexes of Cyclopentadienylnickel with Heterocyclic Carbene. Russ. J. Coord. Chem. 2018, 44, 647–652. [Google Scholar] [CrossRef]
- Ahlrichs, R.; Bar, M.; Haser, M.; Horn, H.; Kolmel, C. Electronic structure calculations on workstation computers: The program system turbomole. Chem. Phys. Lett. 1989, 162, 165–169. [Google Scholar] [CrossRef]
- Adamo, C.; Barone, V. Toward reliable density functional methods without adjustable parameters: The PBE0 model. J. Chem. Phys. 1999, 110, 6158–6169. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef] [PubMed]
- Weigend, F. Accurate Coulomb-fitting basis sets for H to Rn. Phys. Chem. Chem. Phys. 2006, 8, 1057–1065. [Google Scholar] [CrossRef] [PubMed]
- Bader, R.F.W. A quantum theory of molecular structure and its applications. Chem. Rev. 1991, 91, 893–928. [Google Scholar] [CrossRef]
- Contreras-García, J.; Johnson, E.R.; Keinan, S.; Chaudret, R.; Piquemal, J.-P.; Beratan, D.N.; Yang, W. NCIPLOT: A program for plotting non-covalent interaction regions. J. Chem. Theory Comput. 2011, 7, 625–632. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Lu, T.; Chen, F. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef]
- Glendening, E.D.; Landis, C.R.; Weinhold, F. Natural bond orbital methods. WIREs Comput. Mol. Sci. 2012, 2, 1–42. [Google Scholar] [CrossRef]
- Glendening, E.D.; Badenhoop, J.K.; Reed, A.E.; Carpenter, J.E.; Bohmann, J.A.; Morales, C.M.; Karafiloglou, P.; Landis, C.R.; Weinhold, F. NBO 7.0: New Vistas in Localized and Delocalized Chemical Bonding Theory; Theoretical Chemistry Institute, University of Wisconsin-Madison: Madison, WI, USA, 2018. [Google Scholar]
- Kitaura, K.; Morokuma, K. A new energy decomposition scheme for molecular interactions within the Hartree-Fock approximation. Int. J. Quantum Chem. 1976, 10, 325–340. [Google Scholar] [CrossRef]
- Batsanov, S.S. Van der Waals Radii of Elements. Inorg. Mat. 2001, 37, 871–885. [Google Scholar] [CrossRef]
- Ivanov, D.M.; Bokach, N.A.; Kukushkin; Kukushkin, V.Y.; Frontera, A. Metal Centers as Nucleophiles: Oxymoron of Halogen Bond-Involving Crystal Engineering. Chem. Eur. J. 2022, 18, e202103173. [Google Scholar]
- Soldatova, N.S.; Postnikov, P.S.; Ivanov, D.M.; Semyonov, O.V.; Kukurina, O.S.; Guselnikova, O.; Yamauchi, Y.; Wirth, T.; Zhdankin, V.V.; Yusubov, M.S.; et al. Zwitterionic iodonium species afford halogen bond-based porous organic frameworks. Chem. Sci. 2022, 13, 5650–5658. [Google Scholar] [CrossRef] [PubMed]
- Kinzhalov, M.A.; Ivanov, D.M.; Melekhova, A.A.; Bokach, N.A.; Gomila, R.M.; Frontera, A.; Kukushkin, V.Y. Chameleonic metal-bound isocyanides: A π-donating CuI-center imparts nucleophilicity to the isocyanide carbon toward halogen bonding. Inorg. Chem. Front. 2022, 9, 1655–1665. [Google Scholar] [CrossRef]
- Torubaev, Y.V.; Rozhkov, A.V.; Skabitsky, I.V.; Gomila, R.M.; Frontera, A.; Kukushkin, V.Y. Heterovalent chalcogen bonding: Supramolecular assembly driven by the occurrence of a tellurium(II)⋯Ch(I) (Ch = S, Se, Te) linkage. Inorg. Chem. Front. 2022, 9, 5635–5644. [Google Scholar] [CrossRef]
- Artemjev, A.A.; Novikov, A.P.; Burkin, G.M.; Sapronov, A.A.; Kubasov, A.S.; Nenajdenko, V.G.; Khrustalev, V.N.; Borisov, A.V.; Kirichuk, A.A.; Kritchenkov, A.S.; et al. Towards Anion Recognition and Precipitation with Water-Soluble 1,2,4-Selenodiazolium Salts: Combined Structural and Theoretical Study. Int. J. Mol. Sci. 2022, 23, 6372. [Google Scholar] [CrossRef]
- Sapronov, A.A.; Artemjev, A.A.; Burkin, G.M.; Khrustalev, V.N.; Kubasov, A.S.; Nenajdenko, V.G.; Gomila, R.M.; Frontera, A.; Kritchenkov, A.S.; Tskhovrebov, A.G. Robust Supramolecular Dimers Derived from Benzylic-Substituted 1,2,4-Selenodiazolium Salts Featuring Selenium⋯π Chalcogen Bonding. Int. J. Mol. Sci. 2022, 23, 14973. [Google Scholar] [CrossRef]
- Sapronov, A.A.; Kubasov, A.S.; Khrustalev, V.N.; Artemjev, A.A.; Burkin, G.M.; Dukhnovsky, E.A.; Chizhov, A.O.; Kritchenkov, A.S.; Gomila, R.M.; Frontera, A.; et al. Se⋯π Chalcogen Bonding in 1,2,4-Selenodiazolium Tetraphenylborate Complexes. Symmetry 2023, 15, 212. [Google Scholar] [CrossRef]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Burguera, S.; Gomila, R.M.; Bauzá, A.; Frontera, A. Chalcogen Bonds, Halogen Bonds and Halogen···Halogen Contacts in Di- and Tri-iododiorganyltellurium(IV) Derivatives. Inorganics 2023, 11, 209. https://doi.org/10.3390/inorganics11050209
Burguera S, Gomila RM, Bauzá A, Frontera A. Chalcogen Bonds, Halogen Bonds and Halogen···Halogen Contacts in Di- and Tri-iododiorganyltellurium(IV) Derivatives. Inorganics. 2023; 11(5):209. https://doi.org/10.3390/inorganics11050209
Chicago/Turabian StyleBurguera, Sergi, Rosa M. Gomila, Antonio Bauzá, and Antonio Frontera. 2023. "Chalcogen Bonds, Halogen Bonds and Halogen···Halogen Contacts in Di- and Tri-iododiorganyltellurium(IV) Derivatives" Inorganics 11, no. 5: 209. https://doi.org/10.3390/inorganics11050209