
Nickel(II) N-Heterocyclic Carbene Complex for the Hydrogenation of 2-Acetylpyridine under Mild Conditions

 ,
,  , and
, and
Abstract
:1. Introduction
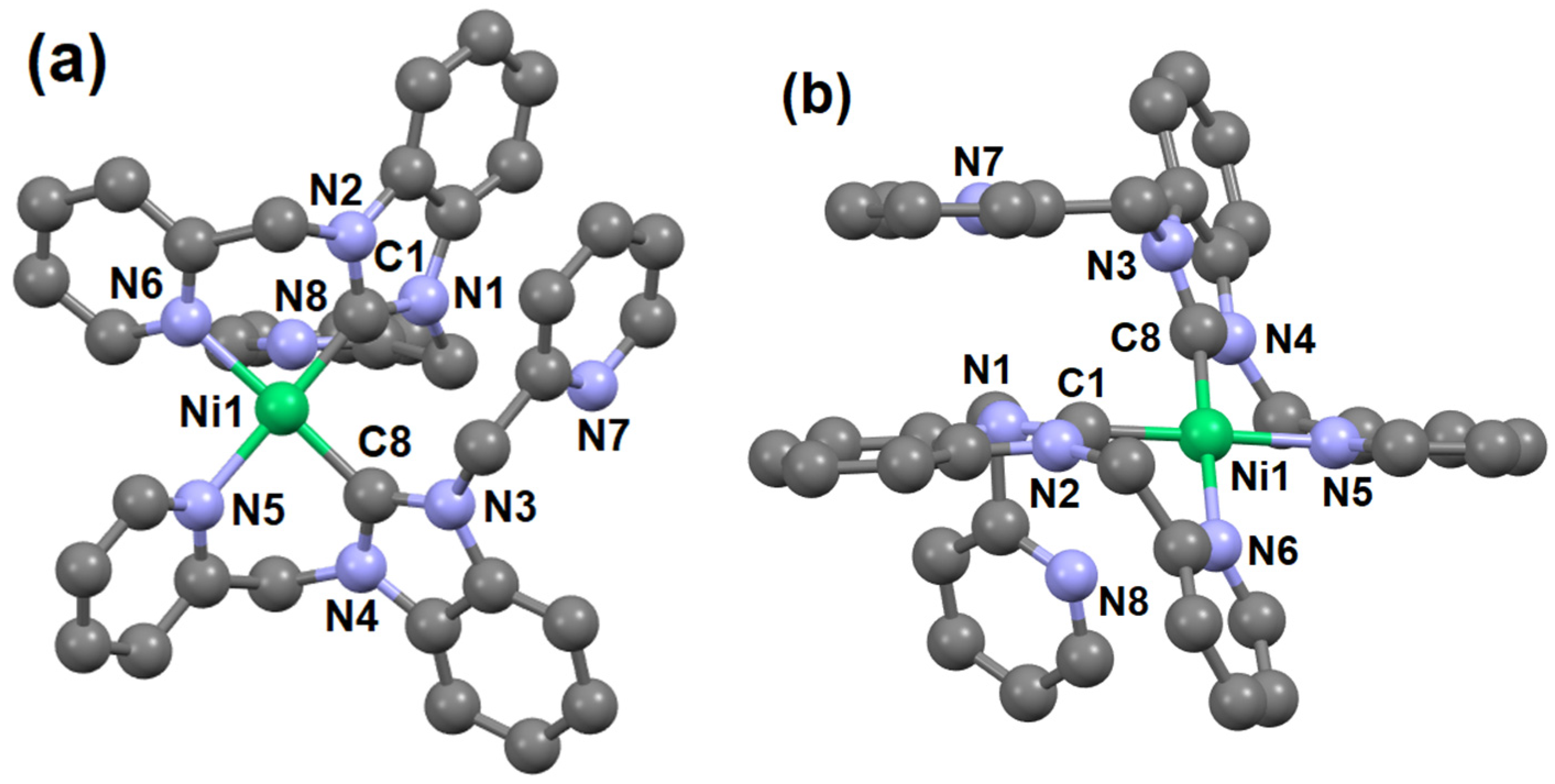
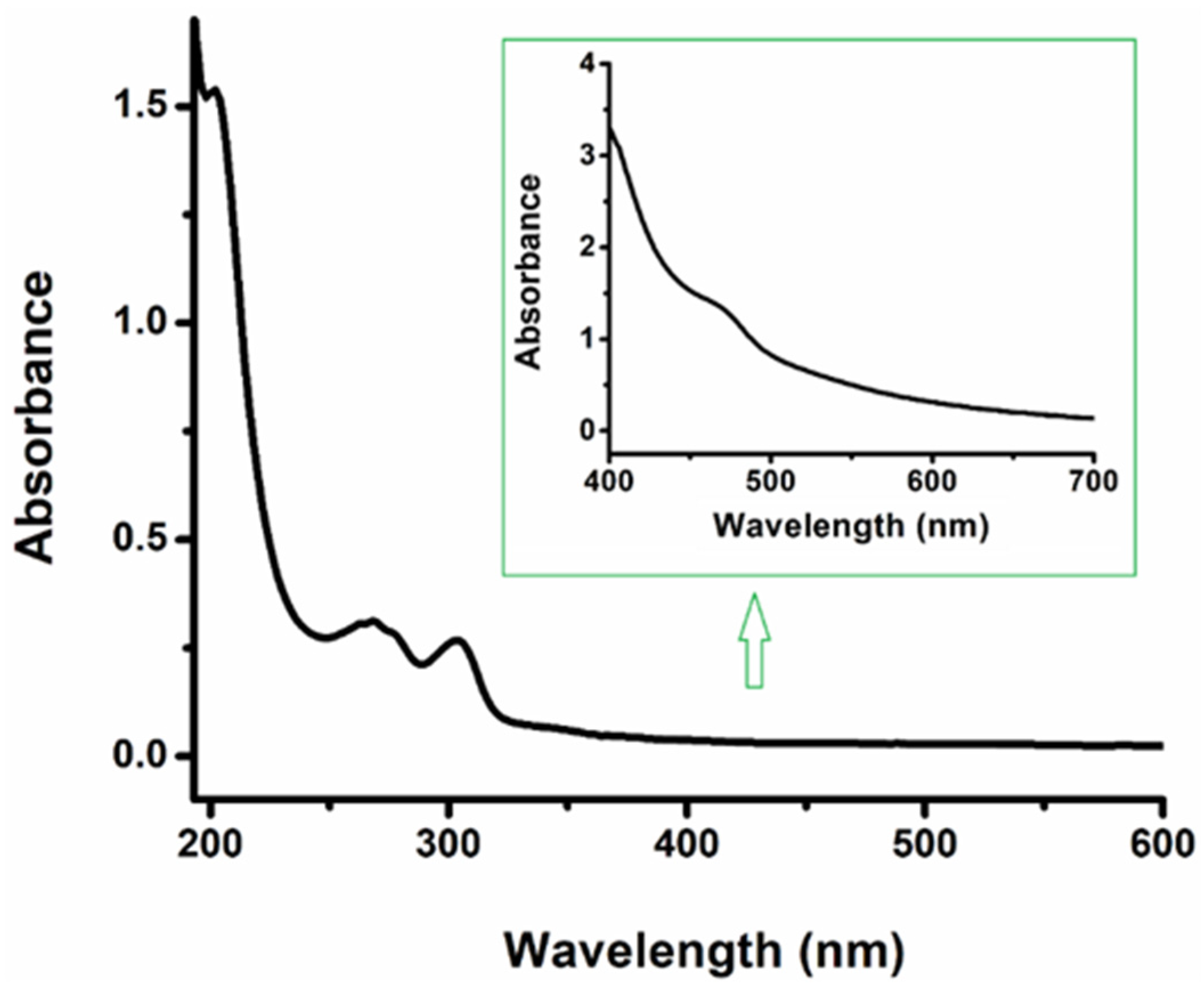
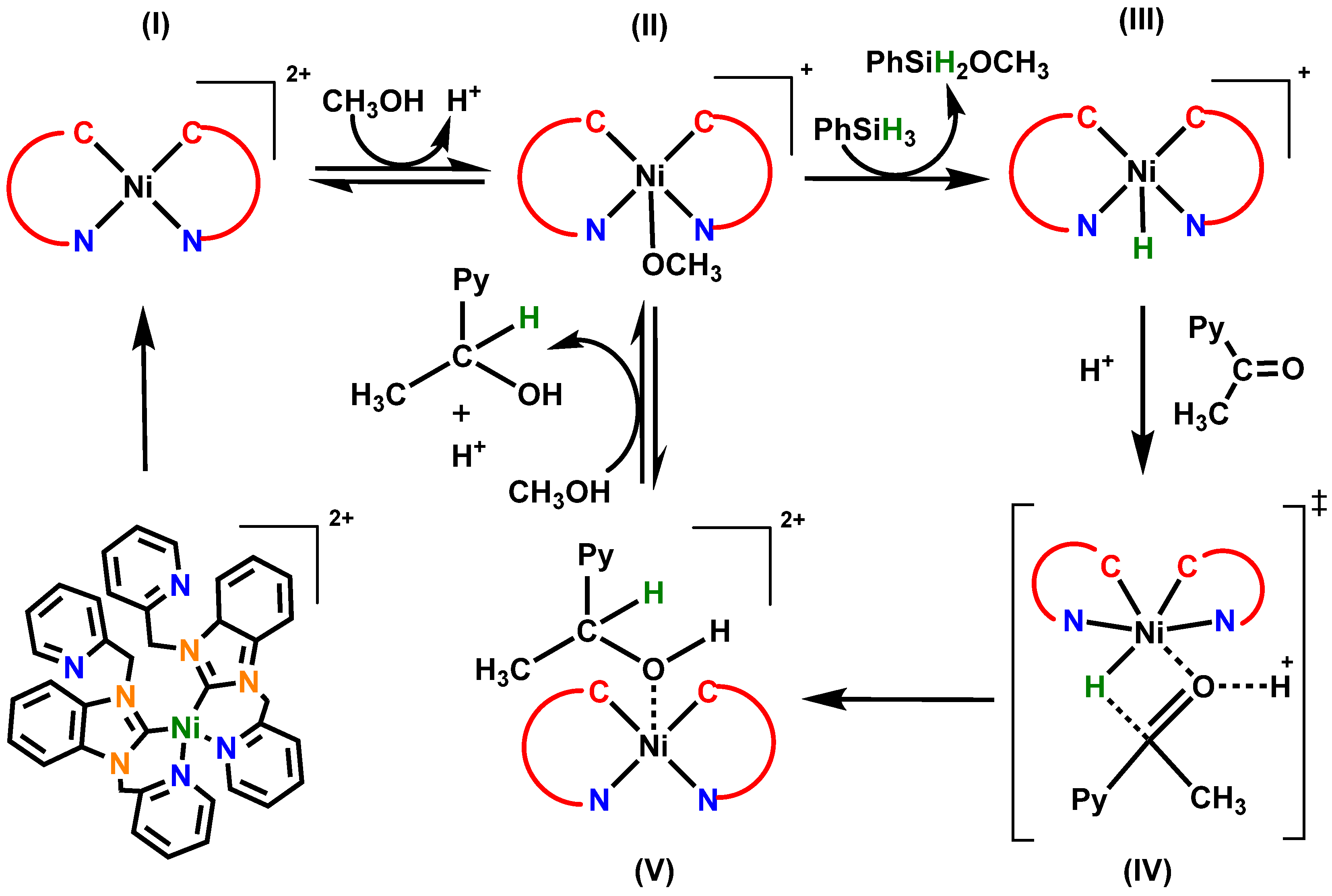
2. Results and Discussions
3. Experimental Section
3.1. Materials
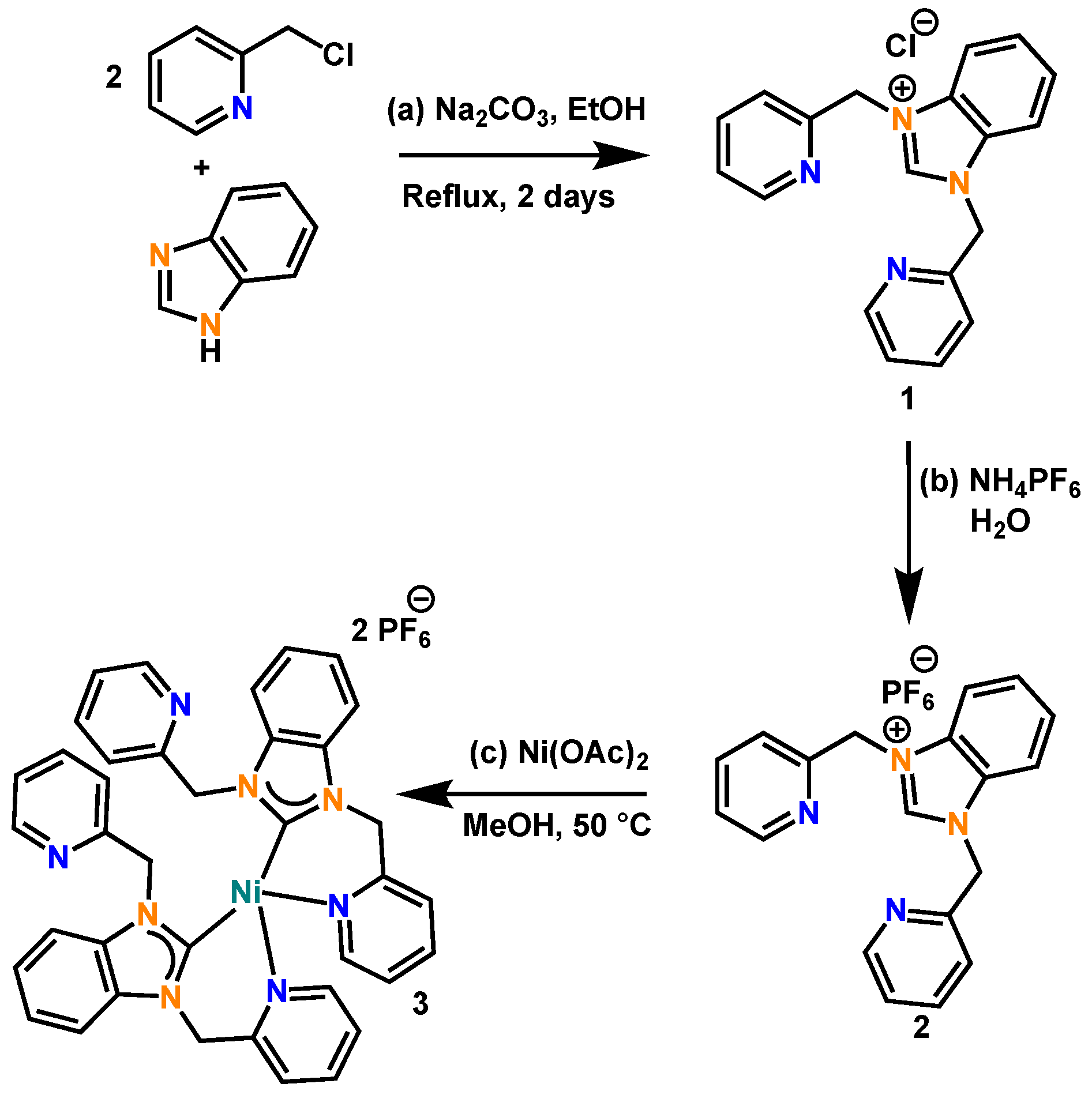
3.2. Synthesis of 1,3-Bis(Pyridin-2-Ylmethyl)-1H-Benzo[d]Imidazol-3-Ium Chloride (1) and Synthesis of 1,3-Bis(Pyridin-2-Ylmethyl)-1H-Benzo[d]Imidazol-3-Ium Hexafluorophosphate (2)
3.3. Synthesis of Complex (3)
3.4. General Procedure for the Hydrogenation of 2-Acetylpyridine
3.5. Experimental Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Johnson, M.T.; Wendt, O.F. Carboxylation reactions involving carbon dioxide insertion into palladium–carbon σ-bonds. J. Organomet. Chem. 2014, 751, 213–220. [Google Scholar] [CrossRef]
- Chen, Q.A.; Ye, Z.S.; Duan, Y.; Zhou, Y.G. Homogeneous palladium-catalyzed asymmetric hydrogenation. Chem. Soc. Rev. 2013, 42, 497–511. [Google Scholar] [CrossRef] [PubMed]
- Sehnal, P.; Taylor, R.J.; Fairlamb, I.J. Emergence of palladium (IV) chemistry in synthesis and catalysis. Chem. Rev. 2010, 110, 824–889. [Google Scholar] [CrossRef] [PubMed]
- Ren, H.; Li, G.F.; Zhu, B.; Lv, X.D.; Yao, L.S.; Wang, X.L.; Su, Z.M.; Guan, W. How does iridium (III) photocatalyst regulate nickel (II) catalyst in metallaphotoredox-catalyzed C–S cross-coupling? Theoretical and experimental insights. ACS Catal. 2019, 9, 3858–3865. [Google Scholar] [CrossRef]
- Prakasham, A.P.; Ghosh, P. Nickel N-heterocyclic carbene complexes and their utility in homogeneous catalysis. Inorg. Chim. Acta 2015, 431, 61–100. [Google Scholar] [CrossRef]
- Hahn, F.E. Heterocyclic carbenes. Angew. Chem. Int. Ed. 2006, 45, 1348–1352. [Google Scholar] [CrossRef]
- Arduengo, A.J., III; Harlow, R.L.; Kline, M. A stable crystalline carbene. J. Am. Chem. Soc. 1991, 113, 361–363. [Google Scholar] [CrossRef]
- Lappert, M.F. Contributions to the Chemistry of Carbenemetal Chemistry. J. Organomet. Chem. 2005, 690, 5467–5473. [Google Scholar] [CrossRef]
- Sellmann, D.; Prechtel, W.; Knoch, F.; Moll, M. Transition metal Complexes with Sulfur Ligands. 94. Synthesis and Reactivity of Nickel, Palladium, and Platinum Complexes with the Thiolate Carbene Ligand ‘S2C’2-. X-ray Structure Determinations of [Ni- (PMe3)(‘S2C’)], [Ni(PPh3)(‘S2C’)], [Ni(‘SC’)2], [Pt(PMe3)(‘S2C’)], and (‘S2CO’)2. Inorg. Chem. 1993, 32, 538–546. [Google Scholar]
- Danopoulos, A.A.; Simler, T.; Braunstein, P. N-heterocyclic carbene complexes of copper, nickel, and cobalt. Chem. Rev. 2019, 119, 3730–3961. [Google Scholar] [CrossRef] [PubMed]
- Radius, U.; Bickelhaupt, F.M. Bonding of imidazol-2-ylidene ligands in nickel complexes. Organometallics 2008, 27, 3410–3414. [Google Scholar] [CrossRef]
- Das, S.; Pati, S.K. On the mechanism of frustrated Lewis pair catalysed hydrogenation of carbonyl compounds. Chem. Eur. J. 2017, 23, 1078–1085. [Google Scholar] [CrossRef]
- Doucet, H.; Ohkuma, T.; Murata, K.; Yokozawa, T.; Kozawa, M.; Katayama, E.; England, A.F.; Ikariya, T.; Noyori, R. trans-[RuCl2(phosphane)2(1,2-diamine)] and Chiral trans-[RuCl2(diphosphane)(1,2-diamine)]: Shelf-Stable Precatalysts for the Rapid, Productive, and Stereoselective Hydrogenation of Ketones. Angew. Chem. Int. Ed. 1998, 37, 1703–1707. [Google Scholar] [CrossRef]
- Nordin, S.J.; Roth, P.; Tarnai, T.; Alonso, D.A.; Brandt, P.; Andersson, P.G. Remote Dipole Effects as a Means to Accelerate [Ru (amino alcohol)]-Catalyzed Transfer Hydrogenation of Ketones. Chem. Eur. J. 2001, 7, 1431–1436. [Google Scholar] [CrossRef]
- Talwar, D.; Wu, X.; Saidi, O.; Salguero, N.P.; Xiao, J. Versatile iridicycle catalysts for highly efficient and chemoselective transfer hydrogenation of carbonyl compounds in water. Chem. Eur. J. 2014, 20, 12835–12842. [Google Scholar] [CrossRef] [PubMed]
- Marulasiddeshwara, M.B.; Kumar, P.R. Hydrogenation of carbonyl compounds to alcohols catalyzed by lignin supported palladium nanoparticles. Mater. Today Proc. 2019, 9, 295–305. [Google Scholar] [CrossRef]
- Ohkuma, T.; Takeno, H.; Honda, Y.; Noyori, R. Asymmetric Hydrogenation of Ketones with Polymer-Bound BINAP/Diamine Ruthenium Catalysts. Adv. Synth. Catal. 2001, 343, 369–375. [Google Scholar] [CrossRef]
- Yang, H.; Huo, N.; Yang, P.; Pei, H.; Lv, H.; Zhang, X. Rhodium catalyzed asymmetric hydrogenation of 2-pyridine ketones. Org. Lett. 2015, 17, 4144–4147. [Google Scholar] [CrossRef] [PubMed]
- Sharma, M.; Adhikari, B.; Awoyemi, R.F.; Perkins, A.M.; Duckworth, A.K.; Donnadieu, B.; Wipf, D.O.; Stokes, S.L.; Emerson, J.P. Copper (II) NHC Catalyst for the Formation of Phenol from Arylboronic Acid. Chemistry 2022, 4, 560–575. [Google Scholar] [CrossRef]
- Luo, S.; Bruggeman, D.F.; Siegler, M.A.; Bouwman, E. Can pendant pyridyl arm assist the proton delivery in electrocatalysis? Org. Lett. 2018, 477, 24–30. [Google Scholar] [CrossRef]
- Zhang, X.; Liu, B.; Liu, A.; Xie, W.; Chen, W. Steric bulkiness-dependent structural diversity in nickel (II) complexes of N-heterocyclic carbenes: Synthesis and structural characterization of tetra-, penta-, and hexacoordinate nickel complexes. Organometallics 2009, 28, 1336–1349. [Google Scholar] [CrossRef]
- Yang, L.; Powell, D.R.; Houser, R.P. Structural variation in copper (I) complexes with pyridylmethylamide ligands: Structural analysis with a new four-coordinate geometry index, τ 4. Dalton Trans. 2007, 9, 955–964. [Google Scholar] [CrossRef] [PubMed]
- Atli, D.D. Synthesis, characterization and catalytic properties of cationic N-heterocyclic carbene silver complexes. Turk. J. Chem. 2021, 45, 577–584. [Google Scholar] [CrossRef]
- Harder, S.; Brettar, J. Rational design of a well-defined soluble calcium hydride complex. Angew. Chem. Int. Ed. 2006, 45, 3474–3478. [Google Scholar] [CrossRef] [PubMed]
- Maddani, M.R.; Moorthy, S.K.; Prabhu, K.R. Chemoselective reduction of azides catalyzed by molybdenum xanthate by using phenylsilane as the hydride source. Tetrahedron 2010, 66, 329–333. [Google Scholar] [CrossRef]
- Ji, P.; Park, J.; Gu, Y.; Clark, D.S.; Hartwig, J.F. Abiotic reduction of ketones with silanes catalysed by carbonic anhydrase through an enzymatic zinc hydride. Nat. Chem. 2021, 13, 312–318. [Google Scholar] [CrossRef]
- Liu, J.T.; Yang, S.; Tang, W.; Yang, Z.; Xu, J. Iridium-catalyzed efficient reduction of ketones in water with formic acid as a hydride donor at low catalyst loading. Green Chem. 2018, 20, 2118–2124. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Bonds | Bond Lengths (Å) |
|---|---|
| Ni1-C1 | 1.871(7) |
| Ni1-C8 | 1.879(7) |
| Ni1-N5 | 1.951(6) |
| Ni1-N6 | 1.952(6) |
| Bonds | Bond Angles (°) |
| N5-Ni1-N6 | 91.7(3) |
| N6-Ni1-C1 | 87.8(3) |
| C1-Ni1-C8 | 93.8(3) |
| C8-Ni1-N5 | 86.7(3) |
| C8-Ni1-N6 | 177.9(3) |
| N5-Ni1-C1 | 179.1(3) |
 | |||||
|---|---|---|---|---|---|
| Entry | Catalyst Loading (Mol %) | Time (h) | Temperature (°C) | Solvent | % Conversion |
| 1 | 2 | 24 | RT | MeOH | 31 |
| 2 | 4 | 24 | RT | MeOH | 39 |
| 3 | 6 | 24 | RT | MeOH | 40 |
| 4 | 8 | 24 | RT | MeOH | 32 |
| 5 | 10 | 24 | RT | MeOH | 38 |
| 6 | 6 | 24 | 0 | MeOH | 21 |
| 7 | 6 | 24 | 40 | MeOH | 30 |
| 8 | 6 | 24 | 60 | MeOH | 32 |
| 9 | 6 | 24 | RT | EtOH | 9 |
| 10 | 6 | 24 | RT | Propanol | 25 |
| 11 | 6 | 24 | RT | Butanol | 7 |
| 12 | 6 | 24 | RT | ACN | 23 |
| 13 | 6 | 24 | RT | H2O | 5 |
| 14 | 6 | 4 | RT | MeOH | 2 |
| 15 | 6 | 6 | RT | MeOH | 5 |
| 16 | 6 | 8 | RT | MeOH | 6 |
| 17 | 6 | 48 | RT | MeOH | 20 |
| 18 | 6 | 72 | RT | MeOH | 28 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sharma, M.; Perkins, A.M.; Duckworth, A.K.; Rouse, E.J.; Donnadieu, B.; Adhikari, B.; Stokes, S.L.; Emerson, J.P. Nickel(II) N-Heterocyclic Carbene Complex for the Hydrogenation of 2-Acetylpyridine under Mild Conditions. Inorganics 2023, 11, 120. https://doi.org/10.3390/inorganics11030120
Sharma M, Perkins AM, Duckworth AK, Rouse EJ, Donnadieu B, Adhikari B, Stokes SL, Emerson JP. Nickel(II) N-Heterocyclic Carbene Complex for the Hydrogenation of 2-Acetylpyridine under Mild Conditions. Inorganics. 2023; 11(3):120. https://doi.org/10.3390/inorganics11030120
Chicago/Turabian StyleSharma, Mitu, Amanda M. Perkins, Alison K. Duckworth, Emily J. Rouse, Bruno Donnadieu, Bhupendra Adhikari, Sean L. Stokes, and Joseph P. Emerson. 2023. "Nickel(II) N-Heterocyclic Carbene Complex for the Hydrogenation of 2-Acetylpyridine under Mild Conditions" Inorganics 11, no. 3: 120. https://doi.org/10.3390/inorganics11030120