
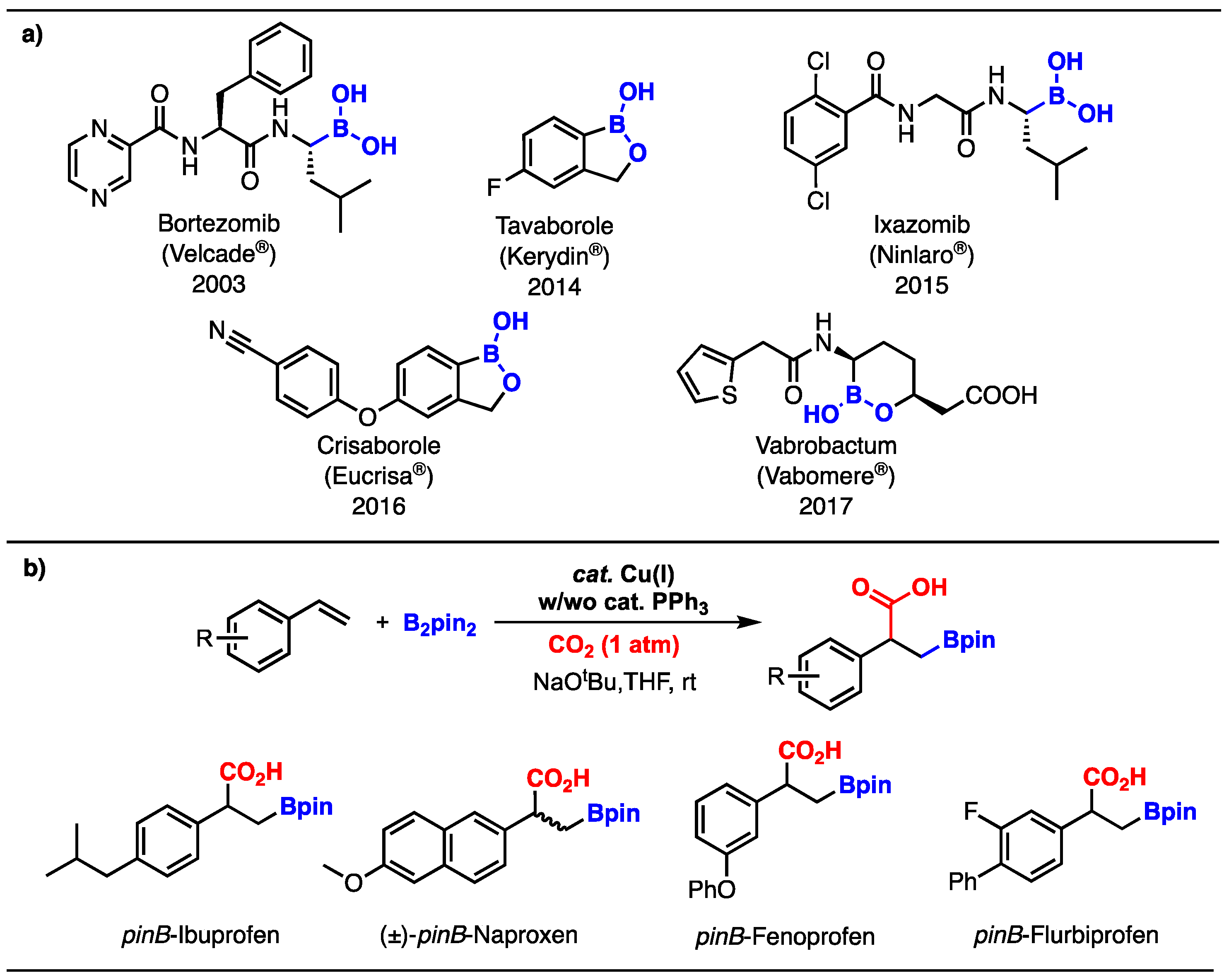
Synthesis of Novel Multifunctional bora-Ibuprofen Derivatives
 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
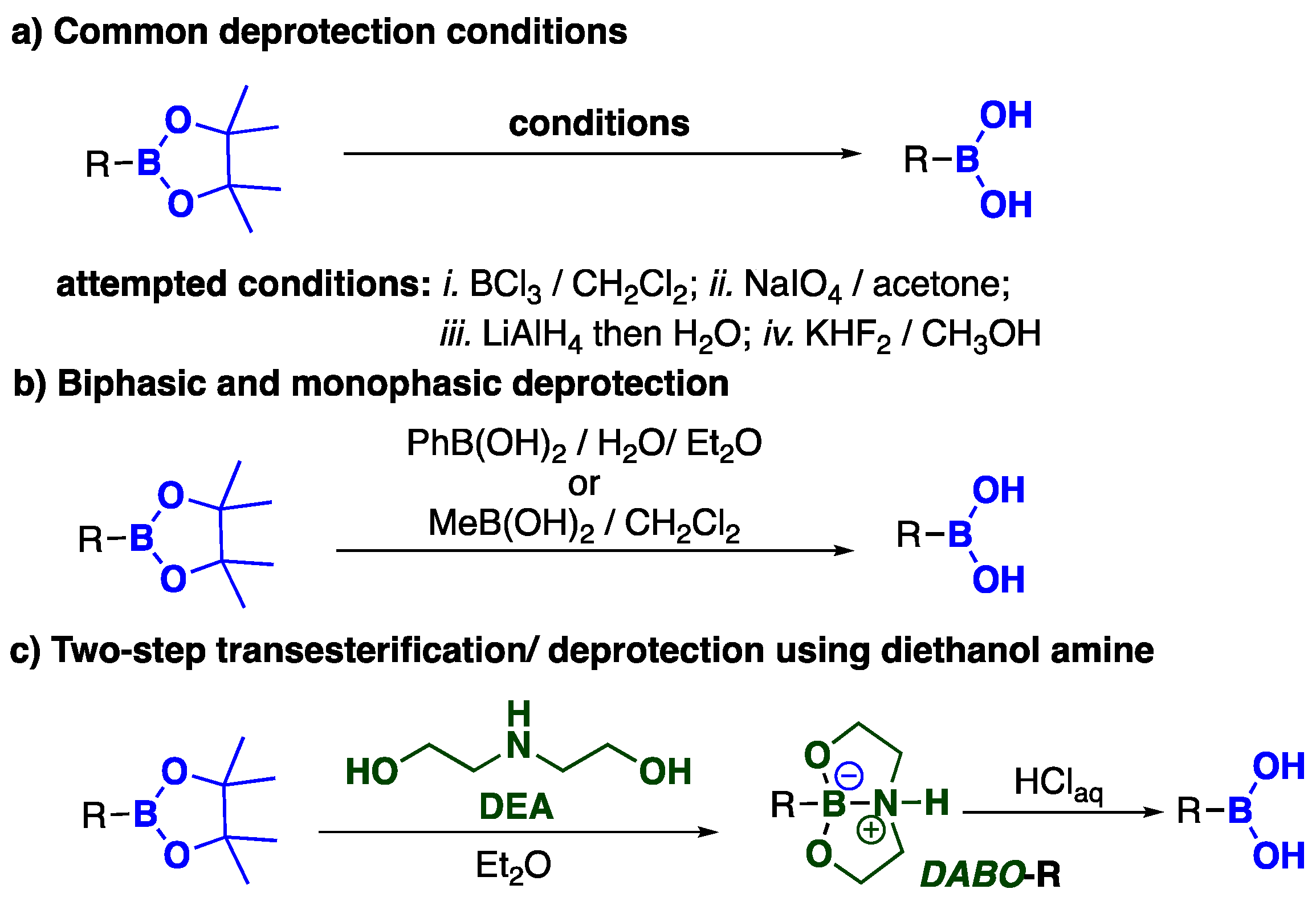
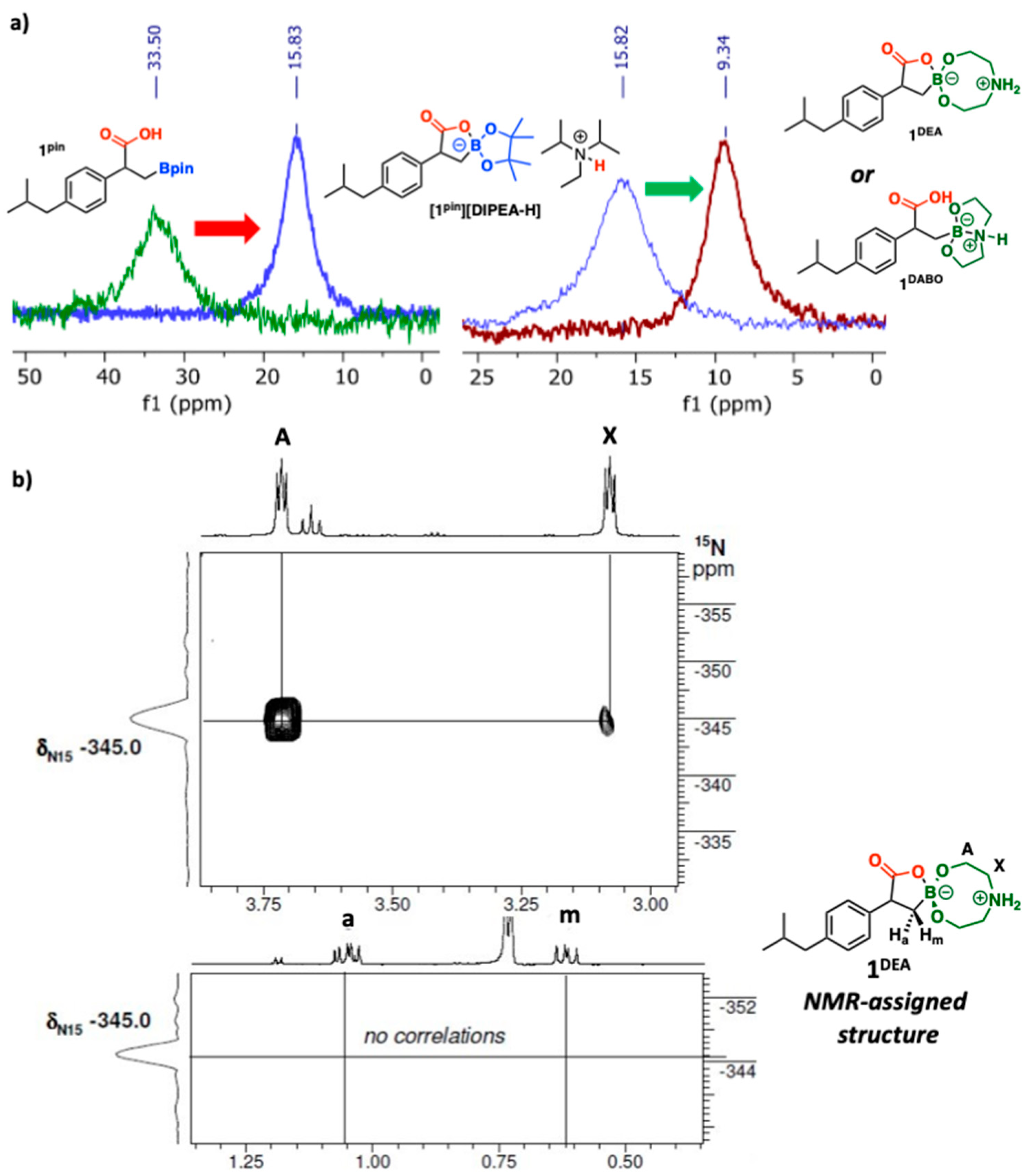
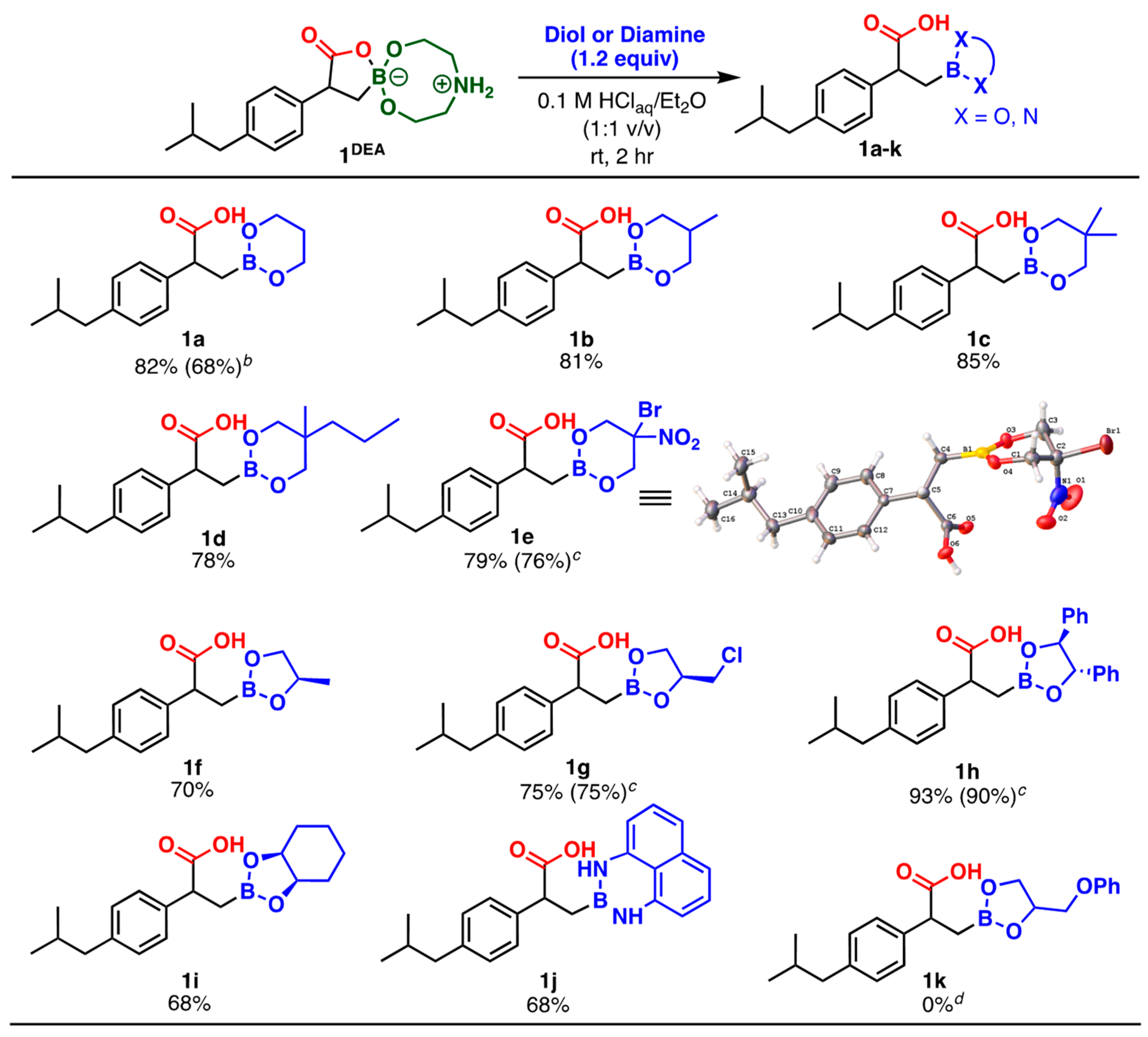
2. Results and Discussion
3. Materials and Methods
3.1. General Methods
3.2. General Procedure for Preparing the Spirocyclic Boralactonate Salt [1pin][DIPEA-H]
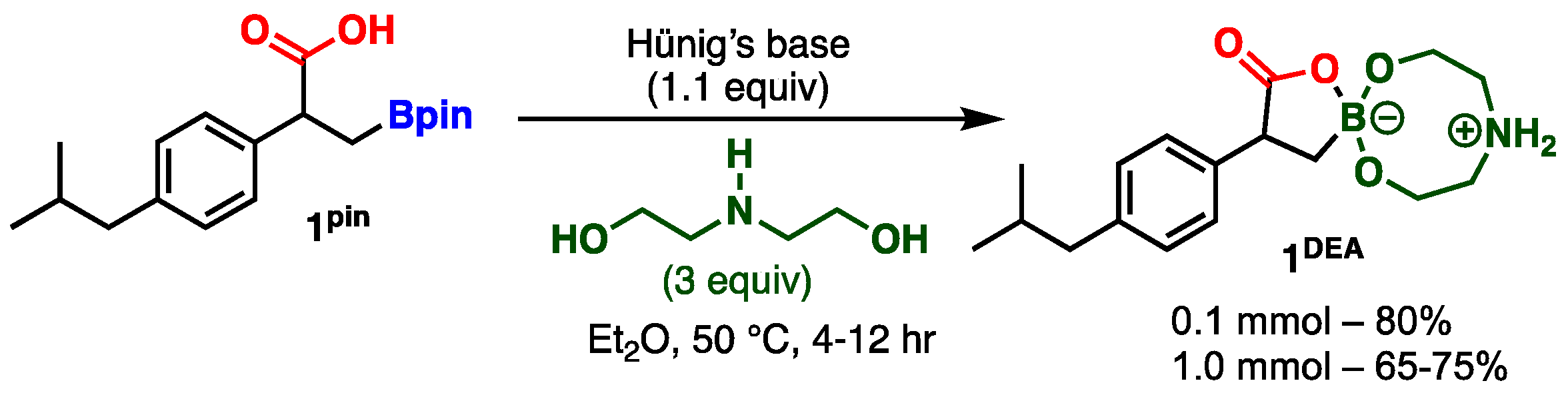
3.3. General Procedure for the Synthesis of Diethanolamine Boronate Ibuprofen 1DEA
3.4. General Procedure for Preparing the bora-Ibuprofen Derivatives 1a–j
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hall, D.G. (Ed.) Boronic Acids: Preparation and Applications in Organic Synthesis, Medicine and Materials, 2nd completely revised ed.; Wiley-VCH: Weinheim, Germany, 2011; ISBN 978-3-527-32598-6. [Google Scholar]
- Miyaura, N.; Suzuki, A. Palladium-Catalyzed Cross-Coupling Reactions of Organoboron Compounds. Chem. Rev. 1995, 95, 2457–2483. [Google Scholar] [CrossRef]
- Chen, J.; Li, J.; Dong, Z. A Review on the Latest Progress of Chan-Lam Coupling Reaction. Adv. Synth. Catal. 2020, 362, 3311–3331. [Google Scholar] [CrossRef]
- Pattison, G. Fluorination of Organoboron Compounds. Org. Biomol. Chem. 2019, 17, 5651–5660. [Google Scholar] [CrossRef] [PubMed]
- Baker, S.J.; Ding, C.Z.; Akama, T.; Zhang, Y.-K.; Hernandez, V.; Xia, Y. Therapeutic Potential of Boron-Containing Compounds. Future Med. Chem. 2009, 1, 1275–1288. [Google Scholar] [CrossRef]
- Fernandes, G.F.S.; Denny, W.A.; Dos Santos, J.L. Boron in Drug Design: Recent Advances in the Development of New Therapeutic Agents. Eur. J. Med. Chem. 2019, 179, 791–804. [Google Scholar] [CrossRef] [PubMed]
- Das, B.C.; Nandwana, N.K.; Das, S.; Nandwana, V.; Shareef, M.A.; Das, Y.; Saito, M.; Weiss, L.M.; Almaguel, F.; Hosmane, N.S.; et al. Boron Chemicals in Drug Discovery and Development: Synthesis and Medicinal Perspective. Molecules 2022, 27, 2615. [Google Scholar] [CrossRef]
- Das, B.C.; Adil Shareef, M.; Das, S.; Nandwana, N.K.; Das, Y.; Saito, M.; Weiss, L.M. Boron-Containing Heterocycles as Promising Pharmacological Agents. Bioorganic Med. Chem. 2022, 63, 116748. [Google Scholar] [CrossRef]
- Soriano-Ursúa, M.A.; Das, B.C.; Trujillo-Ferrara, J.G. Boron-Containing Compounds: Chemico-Biological Properties and Expanding Medicinal Potential in Prevention, Diagnosis and Therapy. Expert Opin. Ther. Pat. 2014, 24, 485–500. [Google Scholar] [CrossRef]
- Romero-Aguilar, K.S.; Arciniega-Martínez, I.M.; Farfán-García, E.D.; Campos-Rodríguez, R.; Reséndiz-Albor, A.A.; Soriano-Ursúa, M.A. Effects of Boron-Containing Compounds on Immune Responses: Review and Patenting Trends. Expert Opin. Ther. Pat. 2019, 29, 339–351. [Google Scholar] [CrossRef]
- Barrón-González, M.; Montes-Aparicio, A.V.; Cuevas-Galindo, M.E.; Orozco-Suárez, S.; Barrientos, R.; Alatorre, A.; Querejeta, E.; Trujillo-Ferrara, J.G.; Farfán-García, E.D.; Soriano-Ursúa, M.A. Boron-Containing Compounds on Neurons: Actions and Potential Applications for Treating Neurodegenerative Diseases. J. Inorg. Biochem. 2023, 238, 112027. [Google Scholar] [CrossRef]
- Leśnikowski, Z.J. Recent Developments with Boron as a Platform for Novel Drug Design. Expert Opin. Drug Discov. 2016, 11, 569–578. [Google Scholar] [CrossRef] [PubMed]
- Song, S.; Gao, P.; Sun, L.; Kang, D.; Kongsted, J.; Poongavanam, V.; Zhan, P.; Liu, X. Recent Developments in the Medicinal Chemistry of Single Boron Atom-Containing Compounds. Acta Pharm. Sin. B 2021, 11, 3035–3059. [Google Scholar] [CrossRef] [PubMed]
- Butcher, T.W.; McClain, E.J.; Hamilton, T.G.; Perrone, T.M.; Kroner, K.M.; Donohoe, G.C.; Akhmedov, N.G.; Petersen, J.L.; Popp, B.V. Regioselective Copper-Catalyzed Boracarboxylation of Vinyl Arenes. Org. Lett. 2016, 18, 6428–6431. [Google Scholar] [CrossRef]
- Perrone, T.M.; Gregory, A.S.; Knowlden, S.W.; Ziemer, N.R.; Alsulami, R.N.; Petersen, J.L.; Popp, B.V. Beneficial Effect of a Secondary Ligand on the Catalytic Difunctionalization of Vinyl Arenes with Boron and CO2. ChemCatChem 2019, 11, 5814–5820. [Google Scholar] [CrossRef]
- Baughman, N.N.; Akhmedov, N.G.; Petersen, J.L.; Popp, B.V. Experimental and Computational Analysis of CO2 Addition Reactions Relevant to Copper-Catalyzed Boracarboxylation of Vinyl Arenes: Evidence for a Phosphine-Promoted Mechanism. Organometallics 2021, 40, 23–37. [Google Scholar] [CrossRef]
- Kinder, D.H.; Katzenellenbogen, J.A. Acylamido Boronic Acids and Difluoroborane Analogs of Amino Acids: Potent Inhibitors of Chymotrypsin and Elastase. J. Med. Chem. 1985, 28, 1917–1925. [Google Scholar] [CrossRef]
- Martichonok, V.; Jones, J.B. Probing the Specificity of the Serine Proteases Subtilisin Carlsberg and R-Chymotrypsin with Enantiomeric 1-Acetamido Boronic Acids. An Unexpected Reversal of the Normal “L”-Stereoselectivity Preference. J. Am. Chem. Soc. 1996, 118, 950–958. [Google Scholar] [CrossRef]
- Diemer, V.; Chaumeil, H.; Defoin, A.; Carré, C. Syntheses of Extreme Sterically Hindered 4-Methoxyboronic Acids. Tetrahedron 2010, 66, 918–929. [Google Scholar] [CrossRef]
- Coutts, S.J.; Adams, J.; Krolikowski, D.; Snow, R.J. Two Efficient Methods for the Cleavage of Pinanediol Boronate Esters Yielding the Free Boronic Acids. Tetrahedron Lett. 1994, 35, 5109–5112. [Google Scholar] [CrossRef]
- Brown, H.C.; Rangaishenvi, M.V. Organoboranes: LI. Convenient Procedures for the Recovery of Pinanediol in Asymmetric Synthesis via One-carbon Homologation of Boronic Esters. J. Organomet. Chem. 1988, 358, 15–30. [Google Scholar] [CrossRef]
- Molander, G.A.; Ito, T. Cross-Coupling Reactions of Potassium Alkyltrifluoroborates with Aryl and 1-Alkenyl Trifluoromethanesulfonates. Org. Lett. 2001, 3, 393–396. [Google Scholar] [CrossRef] [PubMed]
- Yuen, A.K.L.; Hutton, C.A. Deprotection of Pinacolyl Boronate Esters via Hydrolysis of Intermediate Potassium Trifluoroborates. Tetrahedron Lett. 2005, 46, 7899–7903. [Google Scholar] [CrossRef]
- Inglis, S.R.; Woon, E.C.Y.; Thompson, A.L.; Schofield, C.J. Observations on the Deprotection of Pinanediol and Pinacol Boronate Esters via Fluorinated Intermediates. J. Org. Chem. 2010, 75, 468–471. [Google Scholar] [CrossRef] [PubMed]
- Murphy, J.M.; Tzschucke, C.C.; Hartwig, J.F. One-Pot Synthesis of Arylboronic Acids and Aryl Trifluoroborates by Ir-Catalyzed Borylation of Arenes. Org. Lett. 2007, 9, 757–760. [Google Scholar] [CrossRef] [PubMed]
- Abeysinghe, R.T. Multifunctional Organoboron Compounds and Boralactonate Salts. Ph.D. Dissertation, West Virginia University, Morgantown, WV, USA, 2022. [Google Scholar]
- Snow, R.J.; Bachovchin, W.W.; Barton, R.W.; Campbell, S.J.; Coutts, S.J.; Freeman, D.M.; Gutheil, W.G.; Kelly, T.A.; Kennedy, C.A. Studies on Proline Boronic Acid Dipeptide Inhibitors of Dipeptidyl Peptidase IV: Identification of a Cyclic Species Containing a B-N Bond. J. Am. Chem. Soc. 1994, 116, 10860–10869. [Google Scholar] [CrossRef]
- Pennington, T.E.; Kardiman, C.; Hutton, C.A. Deprotection of Pinacolyl Boronate Esters by Transesterification with Polystyrene–Boronic Acid. Tetrahedron Lett. 2004, 45, 6657–6660. [Google Scholar] [CrossRef]
- Hinkes, S.P.A.; Klein, C.D.P. Virtues of Volatility: A Facile Transesterification Approach to Boronic Acids. Org. Lett. 2019, 21, 3048–3052. [Google Scholar] [CrossRef]
- Reilly, M.; Rychnovsky, S. DABO Boronates: Stable Heterocyclic Boronic Acid Complexes for Use in Suzuki-Miyaura Cross-Coupling Reactions. Synlett 2011, 2011, 2392–2396. [Google Scholar] [CrossRef]
- Bonin, H.; Delacroix, T.; Gras, E. Dioxazaborocanes: Old Adducts, New Tricks. Org. Biomol. Chem. 2011, 9, 4714. [Google Scholar] [CrossRef]
- Song, Y.-L.; Morin, C. Cedranediolborane as a Borylating Agent for the Preparation of Boronic Acids: Synthesis of a Boronated Nucleoside Analogue. Synlett 2001, 2001, 0266–0268. [Google Scholar] [CrossRef]
- Jung, M.E.; Lazarova, T.I. New Efficient Method for the Total Synthesis of (S, S)-Isodityrosine from Natural Amino Acids. J. Org. Chem. 1999, 64, 2976–2977. [Google Scholar] [CrossRef]
- Perttu, E.K.; Arnold, M.; Iovine, P.M. The Synthesis and Characterization of Phenylacetylene Tripodal Compounds Containing Boroxine Cores. Tetrahedron Lett. 2005, 46, 8753–8756. [Google Scholar] [CrossRef]
- Sun, J.; Perfetti, M.T.; Santos, W.L. A Method for the Deprotection of Alkylpinacolyl Boronate Esters. J. Org. Chem. 2011, 76, 3571–3575. [Google Scholar] [CrossRef] [PubMed]
- Inglesby, P.A.; Agnew, L.R.; Carter, H.L.; Ring, O.T. Diethanolamine Boronic Esters: Development of a Simple and Standard Process for Boronic Ester Synthesis. Org. Process Res. Dev. 2020, 24, 1683–1689. [Google Scholar] [CrossRef]
- Doidge-Harrison, S.M.S.V.; Musgrave, O.C.; Wardell, J.L. Structure of tetrahydro-2-naphthyl-4H-1,3,6,2-dioxazaboracine. J. Chem. Crystallogr. 1998, 28, 361–366. [Google Scholar] [CrossRef]
- Kline, M.; Cheatham, S. A Robust Method for Determining 1H-15N Long-Range Correlations: 15N Optimized CIGAR-HMBC Experiments. Magn. Reson. Chem. 2003, 41, 307–314. [Google Scholar] [CrossRef]
- Knowlden, S.W.; Abeysinghe, R.T.; Swistok, A.S.; Ravenscroft, A.C.; Popp, B.V. Synthesis of a Borylated Ibuprofen Derivative through Suzuki Cross-Coupling and Alkene Boracarboxylation Reactions. J. Vis. Exp. 2022, 189, e64571. [Google Scholar] [CrossRef] [PubMed]
- Roest, P.C.; Michel, N.W.M.; Batey, R.A. DABO Boronate Promoted Conjugate Allylation of α,β-Unsaturated Aldehydes Using Copper(II) Catalysis. J. Org. Chem. 2016, 81, 6774–6778. [Google Scholar] [CrossRef]
- Cashman, J.N. The Mechanisms of Action of NSAIDs in Analgesia. Drugs 1996, 52, 13–23. [Google Scholar] [CrossRef]
- Rainsford, K.D. Ibuprofen: Pharmacology, Efficacy and Safety. Inflammopharmacology 2009, 17, 275–342. [Google Scholar] [CrossRef]
- Varrassi, G.; Pergolizzi, J.V.; Dowling, P.; Paladini, A. Ibuprofen Safety at the Golden Anniversary: Are All NSAIDs the Same? A Narrative Review. Adv. Ther. 2020, 37, 61–82. [Google Scholar] [CrossRef]
- Wolfe, M.M.; Lichtenstein, D.R.; Singh, G. Gastrointestinal Toxicity of Nonsteroidal Antiinflammatory Drugs. N. Engl. J. Med. 1999, 340, 1888–1899. [Google Scholar] [CrossRef] [PubMed]
- Bally, M.; Dendukuri, N.; Rich, B.; Nadeau, L.; Helin-Salmivaara, A.; Garbe, E.; Brophy, J.M. Risk of Acute Myocardial Infarction with NSAIDs in Real World Use: Bayesian Meta-Analysis of Individual Patient Data. BMJ 2017, 357, j1909. [Google Scholar] [CrossRef] [PubMed]
- Bindu, S.; Mazumder, S.; Bandyopadhyay, U. Non-Steroidal Anti-Inflammatory Drugs (NSAIDs) and Organ Damage: A Current Perspective. Biochem. Pharmacol. 2020, 180, 114147. [Google Scholar] [CrossRef] [PubMed]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abeysinghe, R.T.; Ravenscroft, A.C.; Knowlden, S.W.; Akhmedov, N.G.; Dolinar, B.S.; Popp, B.V. Synthesis of Novel Multifunctional bora-Ibuprofen Derivatives. Inorganics 2023, 11, 70. https://doi.org/10.3390/inorganics11020070
Abeysinghe RT, Ravenscroft AC, Knowlden SW, Akhmedov NG, Dolinar BS, Popp BV. Synthesis of Novel Multifunctional bora-Ibuprofen Derivatives. Inorganics. 2023; 11(2):70. https://doi.org/10.3390/inorganics11020070
Chicago/Turabian StyleAbeysinghe, Randika T., Alexis C. Ravenscroft, Steven W. Knowlden, Novruz G. Akhmedov, Brian S. Dolinar, and Brian V. Popp. 2023. "Synthesis of Novel Multifunctional bora-Ibuprofen Derivatives" Inorganics 11, no. 2: 70. https://doi.org/10.3390/inorganics11020070