
New Dual Inhibitors of SARS-CoV-2 Based on Metal Complexes with Schiff-Base 4-Chloro-3-Methyl Phenyl Hydrazine: Synthesis, DFT, Antibacterial Properties and Molecular Docking Studies


Abstract
:1. Introduction
2. Results and Discussion
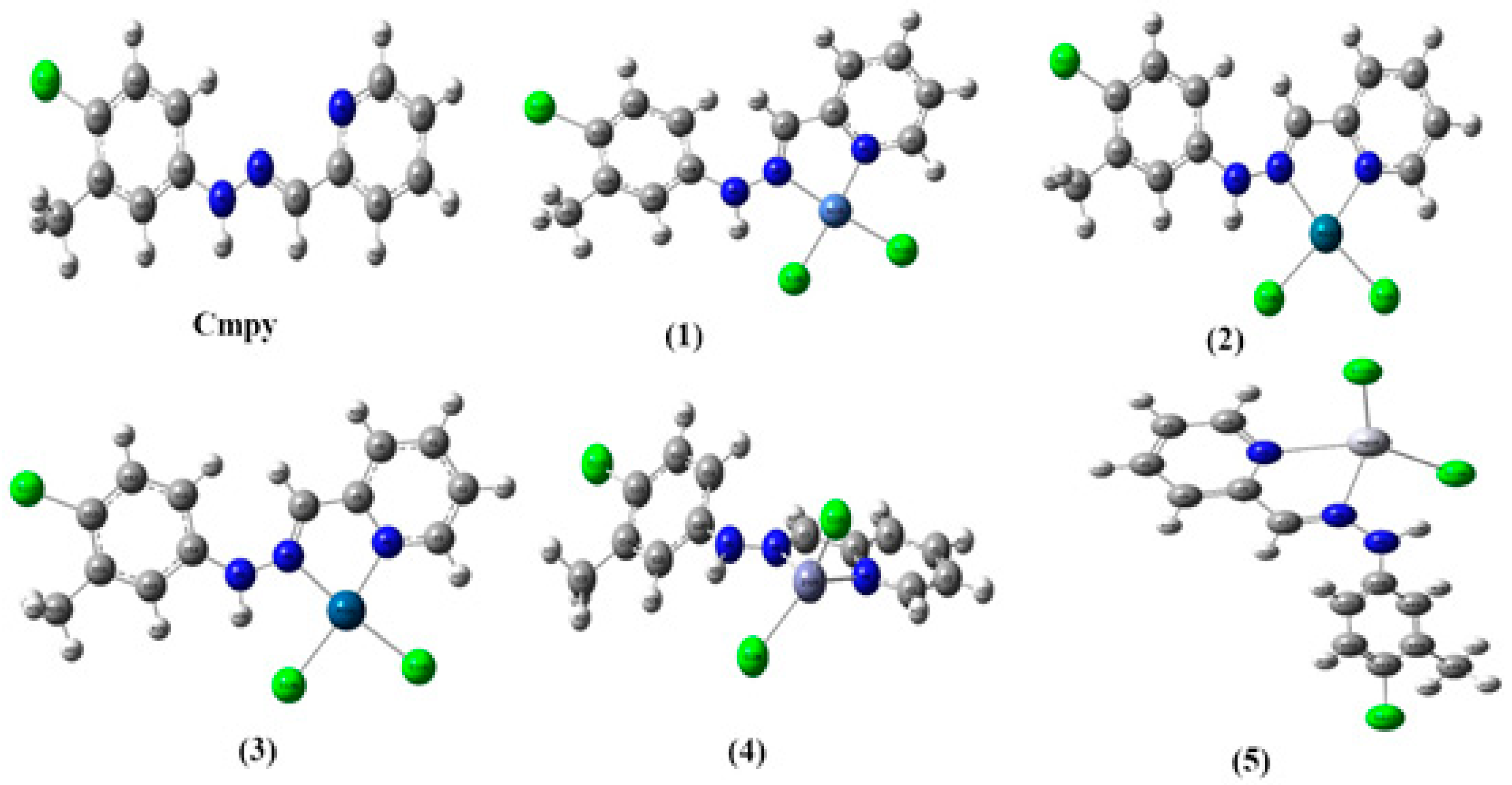
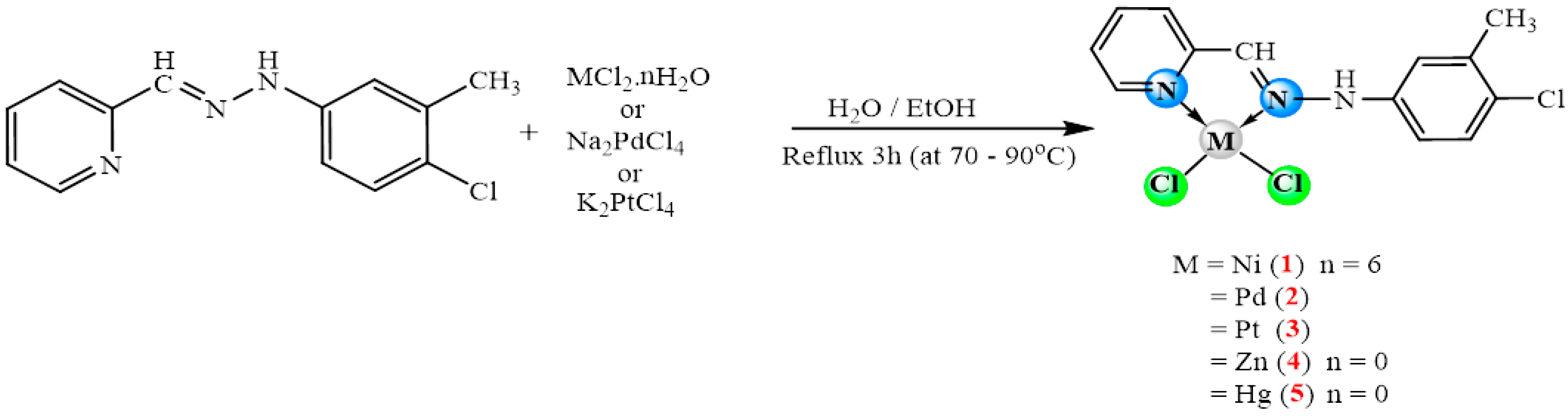
2.1. Synthesis
2.2. IR Spectra
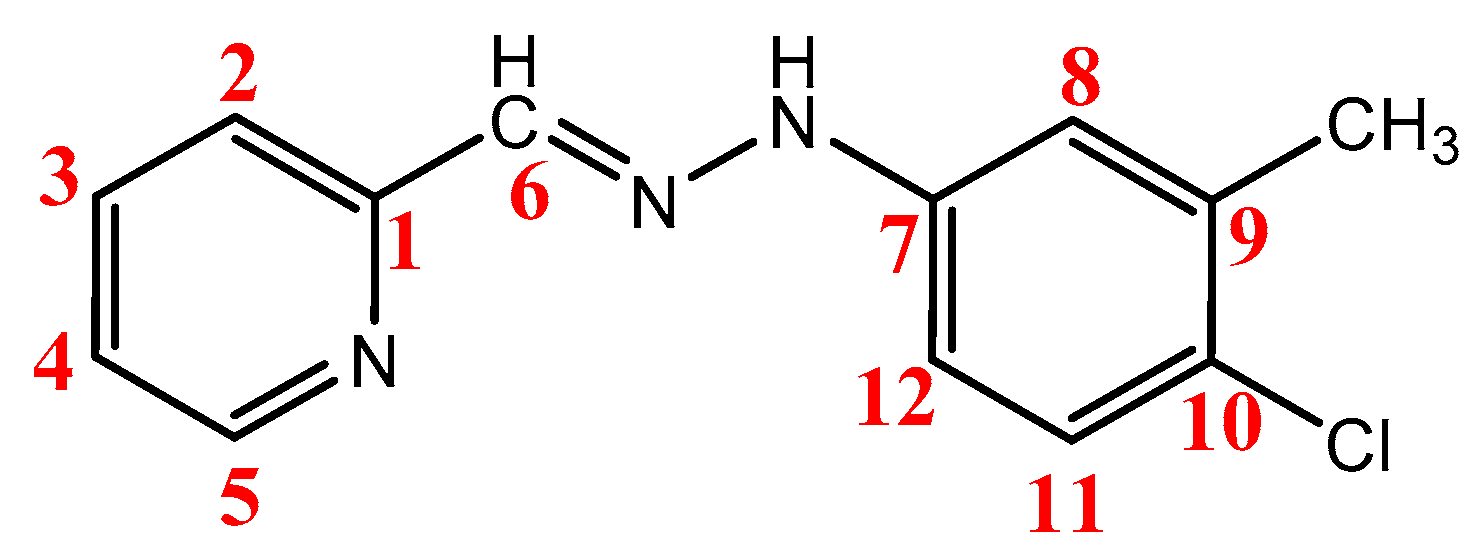
2.3. NMR Spectra
2.4. Antibacterial Studies
2.5. DFT Studies
2.5.1. DFT Calculations Studies
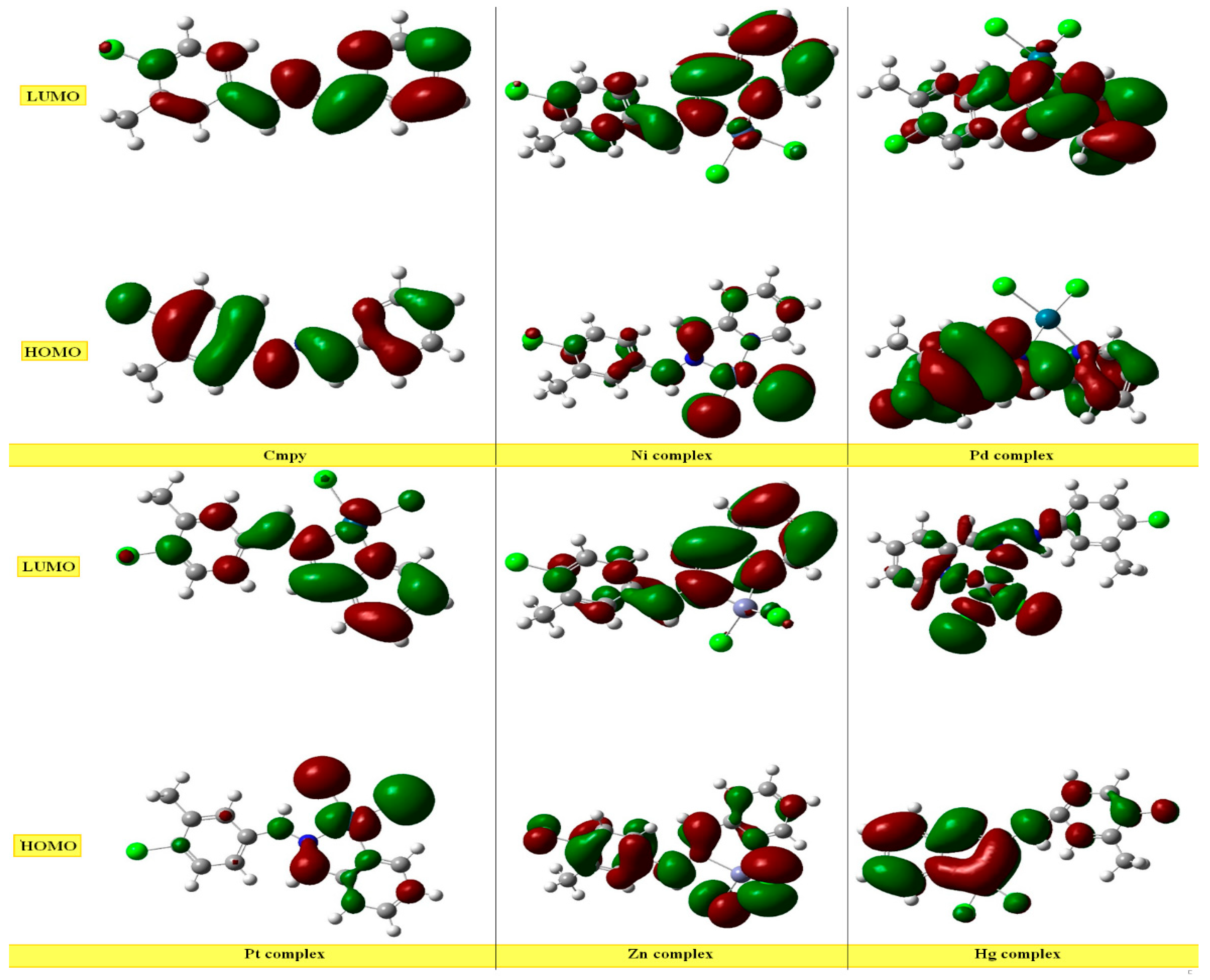
2.5.2. Study of Frontier Orbitals
2.5.3. Chemical Reactivity Descriptors
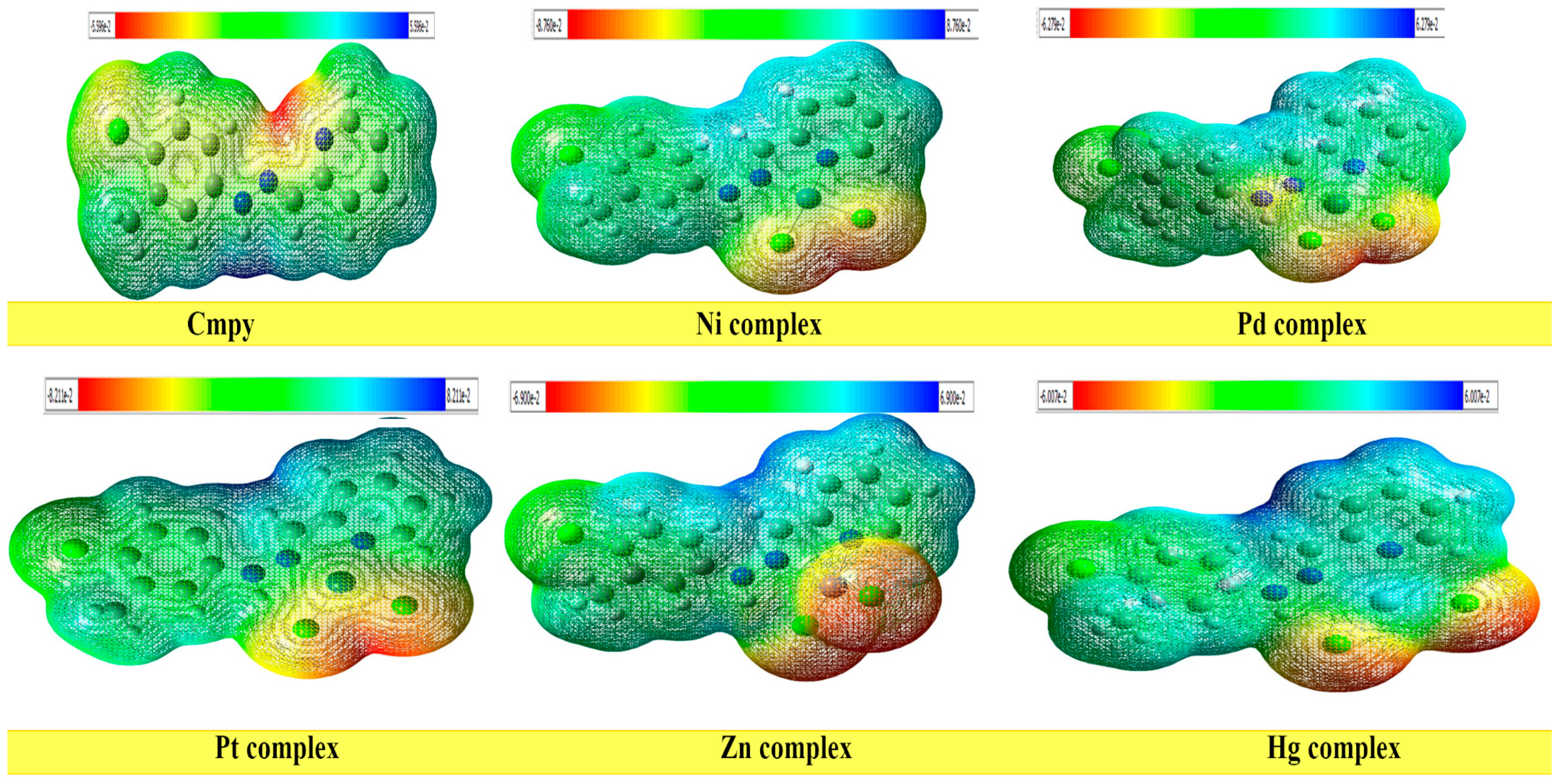
2.5.4. Molecular Electrostatic Potential (MEP)
2.5.5. Mulliken Atomic Charges
2.5.6. Analytical Study of FT-IR spectra
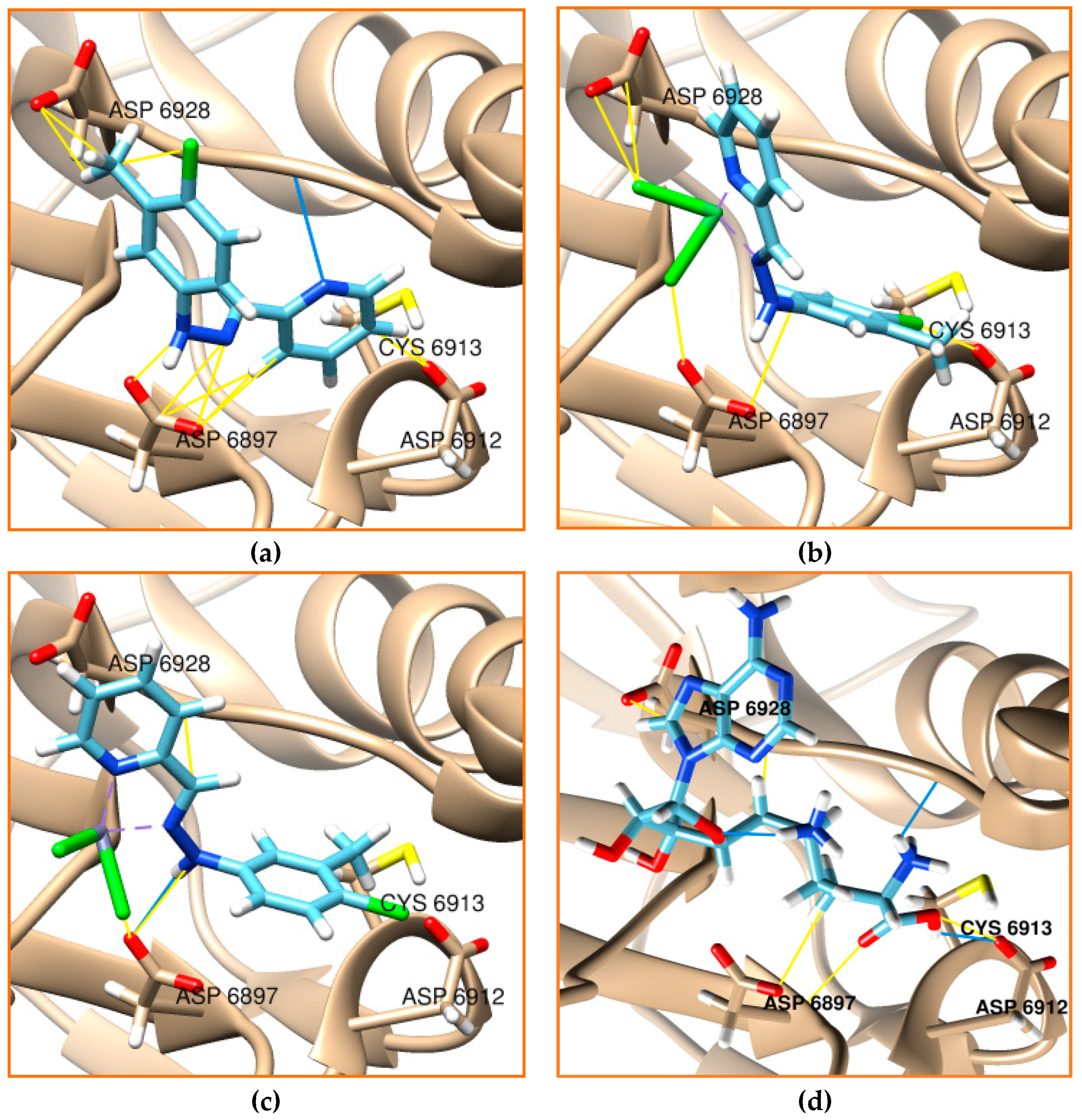
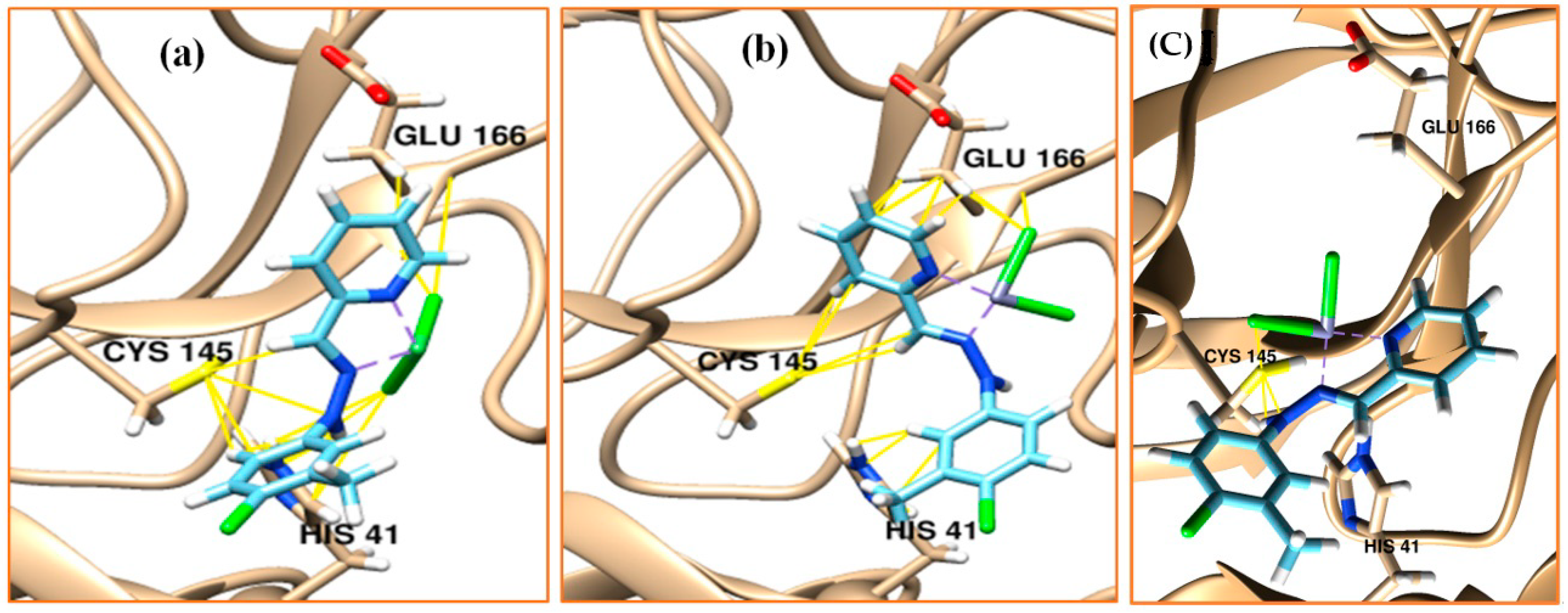
2.6. Molecular Docking
2.7. In Silico ADME Predictions
3. Materials and Methods
3.1. Materials and Instrumental
3.2. Synthesis of Schiff-Base Ligand
3.3. Synthesis of Complex [NiCl2(Cmpy)] (1)
3.4. Antibacterial Studies
3.5. DFT Study
3.6. Molecular Docking
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Turan, N.; Buldurun, K.; Bursal, E.; Mahmoudi, G. Pd (II)-Schiff base complexes: Synthesis, characterization, Suzuki-Miyaura and Mizoroki-Heck cross-coupling reactions, enzyme inhibition and antioxidant activities. J. Organomet. Chem. 2022, 970, 122370. [Google Scholar] [CrossRef]
- Alfonso-Herrera, L.A.; Rosete-Luna, S.; Hernández-Romero, D.; Rivera-Villanueva, J.M.; Olivares-Romero, J.L.; Cruz-Navarro, J.A.; Soto-Contreras, A.; Corona, A.A.; Morales-Morales, D.; Colorado-Peralta, R. Transition metal complexes with tridentate Schiff bases (O N O and O N N) derived from salicylaldehyde: An analysis of their potential anticancer activity. ChemMedChem. 2022, 17, e202200367. [Google Scholar] [CrossRef] [PubMed]
- Fnfoon, D.Y.; Al-Adilee, K.J. Synthesis and spectral characterization of some metal complexes with new heterocyclic azo imidazole dye ligand and study biological activity as anticancer. J. Mol. Struct. 2022, 1271, 134089. [Google Scholar] [CrossRef]
- Ahmetali, E.; Yıldız, B.; Ahi, E.E.; Durmuş, M.; Şener, M.K. Synthesis, Photophysical and Photochemical Properties of Unsymmetrical Zinc (II) Phthalocyanines Bearing 8-Hydroxyquinoline Unit. Polyhedron 2022, 226, 116111. [Google Scholar] [CrossRef]
- Hassan, A.M.; Said, A.O.; Heakal, B.H.; Younis, A.; Aboulthana, W.M.; Mady, M.F. Green Synthesis, Characterization, Antimicrobial and Anticancer Screening of New Metal Complexes Incorporating Schiff Base. ACS Omega 2022, 7, 32418–32431. [Google Scholar] [CrossRef] [PubMed]
- Odularu, A.T. Manganese Schiff Base Complexes, Crystallographic Studies, Anticancer Activities, and Molecular Docking. J. Chem. 2022, 2022, 7062912. [Google Scholar] [CrossRef]
- Frei, A.; Zuegg, J.; Elliott, A.G.; Baker, M.; Braese, S.; Brown, C.; Chen, F.; Dowson, C.G.; Dujardin, G.; Jung, N. Metal complexes as a promising source for new antibiotics. Chem. Sci. 2020, 11, 2627–2639. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Hamon, J.-R. Recent developments in penta-, hexa-and heptadentate Schiff base ligands and their metal complexes. Coord. Chem. Rev. 2019, 389, 94–118. [Google Scholar] [CrossRef]
- Catalano, A.; Sinicropi, M.S.; Iacopetta, D.; Ceramella, J.; Mariconda, A.; Rosano, C.; Scali, E.; Saturnino, C.; Longo, P. A review on the advancements in the field of metal complexes with Schiff bases as antiproliferative agents. Appl. Sci. 2021, 11, 6027. [Google Scholar] [CrossRef]
- Ghanghas, P.; Choudhary, A.; Kumar, D.; Poonia, K. Coordination metal complexes with Schiff bases: Useful pharmacophores with comprehensive biological applications. Inorg. Chem. Commun. 2021, 130, 108710. [Google Scholar] [CrossRef]
- Jamil, W.; Solangi, S.; Ali, M.; Khan, K.M.; Taha, M.; Khuhawar, M.Y. Syntheses, characterization, in vitro antiglycation and DPPH radical scavenging activities of isatin salicylhydrazidehydrazone and its Mn (II), Co (II), Ni (II), Cu (II), and Zn (II) metal complexes. Arab. J. Chem. 2019, 12, 2262–2269. [Google Scholar] [CrossRef] [Green Version]
- El-Tabl, A.S.; Aly, F.A.; Shakdofa, M.M.; Shakdofa, A.M. Synthesis, characterization, and biological activity of metal complexes of azohydrazone ligand. J. Coord. Chem. 2010, 63, 700–712. [Google Scholar] [CrossRef]
- Manimaran, P.; Balasubramaniyan, S.; Azam, M.; Rajadurai, D.; Al-Resayes, S.I.; Mathubala, G.; Manikandan, A.; Muthupandi, S.; Tabassum, Z.; Khan, I. Synthesis, spectral characterization and biological activities of Co (II) and Ni (II) mixed ligand complexes. Molecules 2021, 26, 823. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, A.; Chowdhury, D.; Gomila, R.M.; Chattopadhyay, S. Recent advances on the tetrel bonding interaction in the solid state structure of lead complexes with hydrazine based bis-pyridine Schiff base ligands. Polyhedron 2022, 216, 115670. [Google Scholar] [CrossRef]
- Parvarinezhad, S.; Salehi, M. Synthesis, characterization, anti-proliferative activity and chemistry computation of DFT theoretical methods of hydrazine-based Schiff bases derived from methyl acetoacetate and α-hydroxyacetophenone. J. Mol. Struct. 2021, 1225, 129086. [Google Scholar] [CrossRef]
- Dalapati, S.; Alam, M.A.; Jana, S.; Karmakar, S.; Guchhait, N. “Test kit” for detection of biologically important anions: A salicylidene-hydrazine based Schiff base. Spectrochim. Acta A Mol. Biomol. Spectrosc. 2013, 102, 314–318. [Google Scholar] [CrossRef]
- Jana, S.; Dalapati, S.; Alam, M.A.; Guchhait, N. Spectroscopic, colorimetric and theoretical investigation of Salicylidene hydrazine based reduced Schiff base and its application towards biologically important anions. Spectrochim. Acta A Mol. Biomol. Spectrosc. 2012, 92, 131–136. [Google Scholar] [CrossRef]
- Elkaeed, E.B.; Eissa, I.H.; Elkady, H.; Abdelalim, A.; Alqaisi, A.M.; Alsfouk, A.A.; Elwan, A.; Metwaly, A.M. A Multistage In Silico Study of Natural Potential Inhibitors Targeting SARS-CoV-2 Main Protease. Int. J. Mol. Sci. 2022, 23, 8407. [Google Scholar] [CrossRef]
- Lopes, A.J.O.; Calado, G.P.; Fróes, Y.N.; Araújo, S.A.d.; França, L.M.; Paes, A.M.d.A.; Morais, S.V.d.; Rocha, C.Q.d.; Vasconcelos, C.C. Plant Metabolites as SARS-CoV-2 Inhibitors Candidates: In Silico and In Vitro Studies. Pharmaceuticals 2022, 15, 1045. [Google Scholar] [CrossRef]
- Bhullar, K.S.; Nael, M.A.; Elokely, K.M.; Drews, S.J.; Wu, J. Structurally Modified Bioactive Peptide Inhibits SARS-CoV-2 Lentiviral Particles Expression. Pharmaceutics 2022, 14, 2045. [Google Scholar] [CrossRef]
- Narayanan, A.; Narwal, M.; Majowicz, S.A.; Varricchio, C.; Toner, S.A.; Ballatore, C.; Brancale, A.; Murakami, K.S.; Jose, J. Identification of SARS-CoV-2 inhibitors targeting Mpro and PLpro using in-cell-protease assay. Commun. Biol. 2022, 5, 169. [Google Scholar] [CrossRef] [PubMed]
- Kneller, D.W.; Li, H.; Phillips, G.; Weiss, K.L.; Zhang, Q.; Arnould, M.A.; Jonsson, C.B.; Surendranathan, S.; Parvathareddy, J.; Blakeley, M.P. Covalent narlaprevir-and boceprevir-derived hybrid inhibitors of SARS-CoV-2 main protease. Nat. Commun. 2022, 13, 2268. [Google Scholar] [CrossRef]
- Kisakov, D.N.; Kisakova, L.A.; Borgoyakova, M.B.; Starostina, E.V.; Taranov, O.S.; Ivleva, E.K.; Pyankov, O.V.; Zaykovskaya, A.V.; Shcherbakov, D.N.; Rudometov, A.P.; et al. Optimization of In Vivo Electroporation Conditions and Delivery of DNA Vaccine Encoding SARS-CoV-2 RBD Using the Determined Protocol. Pharmaceutics 2022, 14, 2259. [Google Scholar] [CrossRef] [PubMed]
- Fadlalla, M.; Ahmed, M.; Ali, M.; Elshiekh, A.A.; Yousef, B.A. Molecular docking as a potential approach in repurposing drugs against COVID-19: A systematic review and novel pharmacophore models. Curr. Pharmacol. Rep. 2022, 8, 212–226. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.; Bhardwaj, V.K.; Sharma, J.; Purohit, R.; Kumar, S. In-silico evaluation of bioactive compounds from tea as potential SARS-CoV-2 nonstructural protein 16 inhibitors. J. Tradit. Complement. Med. 2022, 12, 35–43. [Google Scholar] [CrossRef]
- Krafcikova, P.; Silhan, J.; Nencka, R.; Boura, E. Structural analysis of the SARS-CoV-2 methyltransferase complex involved in RNA cap creation bound to sinefungin. Nat. Commun. 2020, 11, 3717. [Google Scholar] [CrossRef]
- Rosas-Lemus, M.; Minasov, G.; Shuvalova, L.; Inniss, N.L.; Kiryukhina, O.; Brunzelle, J.; Satchell, K.J. High-resolution structures of the SARS-CoV-2 2′-O-methyltransferase reveal strategies for structure-based inhibitor design. Sci. Signal. 2020, 13, eabe1202. [Google Scholar] [CrossRef]
- Tweedy, B.G. Plant Extracts with Metal Ions as Potential Antimicrobial Agents. Phytopathology 1964, 55, 910–918. [Google Scholar]
- Thakur, M.L.; Coss, R.; Howell, R.; Vassileva-Belnikolovska, D.; Liu, J.; Rao, S.P.; Spana, G.; Wachsberger, P.; Leeper, D.L. Role of lipid-soluble complexes in targeted tumor therapy. J. Nucl. Med. 2003, 44, 1293–1300. [Google Scholar]
- Lewis, D.; Ioannides, C.; Parke, D. Interaction of a series of nitriles with the alcohol-inducible isoform of P450: Computer analysis of structure—Activity relationships. Xenobiotica 1994, 24, 401–408. [Google Scholar] [CrossRef]
- Shahidha, R.; Muthu, S.; Raja, M.; Muhamed, R.R.; Narayana, B.; Nayak, P.S.; Sarojini, B. Spectroscopic (FT-IR, FT-Raman), first order hyperpolarizabilities, NBO, Fukui function and molecular docking study of N-(4-Chloro-3-methylphenyl)-2-phenylacetamide. Optik 2017, 140, 1127–1142. [Google Scholar] [CrossRef]
- Fukui, K. Role of Frontier Orbitals in Chemical Reactions. Science 1982, 218, 747–754. [Google Scholar] [CrossRef]
- Yang, W.; Parr, R.G. Hardness, Softness, and the Fukui Function in the Electronic Theory of Metals and Catalysis. Proc. Natl. Acad. Sci. USA 1985, 82, 6723–6726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Secretan, P.-H.; Annereau, M.; Kini-Matondo, W.; Prost, B.; Prudhomme, J.; Bournane, L.; Paul, M.; Yagoubi, N.; Sadou-Yayé, H.; Do, B. Unequal Behaviour between Hydrolysable Functions of Nirmatrelvir under Stress Conditions: Structural and Theoretical Approaches in Support of Preformulation Studies. Pharmaceutics 2022, 14, 1720. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.; Schlegel, H.; Scuseria, G.; Robb, M.; Cheeseman, J.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.; et al. Gaussian 09, Revision C.01; Gaussian, Inc.,: Wallingford, CT, USA, 2009. [Google Scholar]
- Shi, L.; Wen, Z.; Song, Y.; Wang, J.; Yu, D. Computational investigation of potent inhibitors against SARS-CoV-2 2′-O-methyltransferase (NSP16): Structure-based pharmacophore modeling, molecular docking, molecular dynamics simulations and binding free energy calculations. J. Mol. Graph. Model. 2022, 117, 108306. [Google Scholar] [CrossRef] [PubMed]
- Amporndanai, K.; Meng, X.; Shang, W.; Jin, Z.; Rogers, M.; Zhao, Y.; Rao, Z.; Liu, Z.J.; Yang, H.; Zhang, L.; et al. Inhibition mechanism of SARS-CoV-2 main protease by ebselen and its derivatives. Nat. Commun. 2021, 12, 3061. [Google Scholar] [CrossRef]
- Bauer, A.W.; PERRY, D.M.; KIRBY, W.M. Single-disk antibiotic-sensitivity testing of staphylococci: An analysis of technique and results. AMA Arch. Intern. Med. 1959, 104, 208–216. [Google Scholar] [CrossRef]
- Mahalapbutr, P.; Kongtaworn, N.; Rungrotmongkol, T. Structural insight into the recognition of S-adenosyl-L-homocysteine and sinefungin in SARS-CoV-2 NSP16/NSP10 RNA cap 2′-O-Methyltransferase. Comput. Struct. Biotechnol. J. 2020, 18, 2757–2765. [Google Scholar] [CrossRef]
- Eissa, I.H.; Alesawy, M.S.; Saleh, A.M.; Elkaeed, E.B.; Alsfouk, B.A.; El-Attar, A.-A.M.; Metwaly, A.M. Ligand and structure-based in silico determination of the most promising SARS-CoV-2 NSP16-NSP10 2′-o-Methyltransferase complex inhibitors among 3009 FDA approved drugs. Molecules 2022, 27, 2287. [Google Scholar] [CrossRef]
- Faletrov, Y.V.; Staravoitava, V.A.; Dudko, A.R.; Shkumatov, V.M. Application of docking-based inverse high throughput virtual screening to found phytochemical covalent inhibitors of SARS-CoV-2 main protease, NSP12 and NSP16. 2022. Available online: https://europepmc.org/article/ppr/ppr473072 (accessed on 17 December 2022).
- Nguyen, H.L.; Thai, N.Q.; Li, M.S. Identifying inhibitors of NSP16-NSP10 of SARS-CoV-2 from large databases. J. Biomol. Struct. Dyn. 2022, 40, 1–10. [Google Scholar] [CrossRef]
- Alves Borges Leal, A.L.; Fonseca Bezerra, C.; Ferreira e Silva, A.K.; Everson da Silva, L.; Bezerra, L.L.; Almeida-Neto, F.W.; Marinho, E.M.; Celedonio Fernandes, C.F.; Nunes da Rocha, M.; Marinho, M.M. Seasonal variation of the composition of essential oils from Piper cernuum Vell and Piper rivinoides Kunth, ADMET study, DFT calculations, molecular docking and dynamics studies of major components as potent inhibitors of the heterodimer methyltransferase complex NSP16-NSP10 SARS COV-2 protein. J. Biomol. Struct. Dyn. 2022, 40, 1–19. [Google Scholar]
- Gomes, J.P.A.; de Oliveira Rocha, L.; Leal, C.E.Y.; de Alencar Filho, E.B. Virtual screening of molecular databases for potential inhibitors of the NSP16/NSP10 methyltransferase from SARS-CoV-2. J. Mol. Struct. 2022, 1261, 132951. [Google Scholar] [CrossRef] [PubMed]
- Bhardwaj, A.; Sharma, S.; Singh, S.K. Molecular docking studies to identify promising natural inhibitors targeting SARS-CoV-2 NSP10-NSP16 protein complex. Turk. J. Pharm. Sci. 2022, 19, 93. [Google Scholar] [CrossRef] [PubMed]
- Mishra, A.; Mulpuru, V.; Mishra, N. Identification of SARS-CoV-2 inhibitors through phylogenetics and drug repurposing. Struct. Chem. 2022, 33, 1789–1797. [Google Scholar] [CrossRef] [PubMed]
- Liang, J.; Pitsillou, E.; Burbury, L.; Hung, A.; Karagiannis, T.C. In silico investigation of potential small molecule inhibitors of the SARS-CoV-2 NSP10-NSP16 methyltransferase complex. Chem. Phys. Lett. 2021, 774, 138618. [Google Scholar] [CrossRef]
- El Hassab, M.A.; Ibrahim, T.M.; Al-Rashood, S.T.; Alharbi, A.; Eskandrani, R.O.; Eldehna, W.M. In silico identification of novel SARS-COV-2 2′-O-methyltransferase (NSP16) inhibitors: Structure-based virtual screening, molecular dynamics simulation and MM-PBSA approaches. J. Enzym. Inhib. Med. Chem. 2021, 36, 727–736. [Google Scholar] [CrossRef]
- Romero-Martínez, B.S.; Montaño, L.M.; Solís-Chagoyán, H.; Sommer, B.; Ramírez-Salinas, G.L.; Pérez-Figueroa, G.E.; Flores-Soto, E. Possible beneficial actions of caffeine in SARS-CoV-2. Int. J. Mol. Sci. 2021, 22, 5460. [Google Scholar] [CrossRef]
- Elzupir, A.O. Caffeine and caffeine-containing pharmaceuticals as promising inhibitors for 3-chymotrypsin-like protease of SARS-CoV-2. J. Biomol. Struct. Dyn. 2022, 40, 2113–2120. [Google Scholar] [CrossRef]
- Rolta, R.; Salaria, D.; Sharma, B.; Awofisayo, O.; Fadare, O.A.; Sharma, S.; Patel, C.N.; Kumar, V.; Sourirajan, A.; Baumler, D.J. Methylxanthines as Potential Inhibitor of SARS-CoV-2: An in Silico Approach. Curr. Pharmacol. Rep. 2022, 8, 149–170. [Google Scholar] [CrossRef]
- Abdallah, H.M.; El-Halawany, A.M.; Sirwi, A.; El-Araby, A.M.; Mohamed, G.A.; Ibrahim, S.R.; Koshak, A.E.; Asfour, H.Z.; Awan, Z.A.; Elfaky, M.A. Repurposing of some natural product isolates as SARS-COV-2 main protease inhibitors via in vitro cell free and cell-based antiviral assessments and molecular modeling approaches. Pharmaceuticals 2021, 14, 213. [Google Scholar] [CrossRef]
- van de Sand, L.; Bormann, M.; Alt, M.; Schipper, L.; Heilingloh, C.S.; Steinmann, E.; Todt, D.; Dittmer, U.; Elsner, C.; Witzke, O. Glycyrrhizin effectively inhibits SARS-CoV-2 replication by inhibiting the viral main protease. Viruses 2021, 13, 609. [Google Scholar] [CrossRef] [PubMed]
- Lockbaum, G.J.; Reyes, A.C.; Lee, J.M.; Tilvawala, R.; Nalivaika, E.A.; Ali, A.; Kurt Yilmaz, N.; Thompson, P.R.; Schiffer, C.A. Crystal structure of SARS-CoV-2 main protease in complex with the non-covalent inhibitor ML188. Viruses 2021, 13, 174. [Google Scholar] [CrossRef] [PubMed]
- Elzupir, A.O. Molecular Docking and Dynamics Investigations for Identifying Potential Inhibitors of the 3-Chymotrypsin-like Protease of SARS-CoV-2: Repurposing of Approved Pyrimidonic Pharmaceuticals for COVID-19 Treatment. Molecules 2021, 26, 7458. [Google Scholar] [CrossRef]
- Gammeltoft, K.A.; Zhou, Y.; Duarte Hernandez, C.R.; Galli, A.; Offersgaard, A.; Costa, R.; Pham, L.V.; Fahnøe, U.; Feng, S.; Scheel, T.K. Hepatitis C virus protease inhibitors show differential efficacy and interactions with remdesivir for treatment of SARS-CoV-2 in vitro. Antimicrob. Agents Chemother. 2021, 65, e0268020. [Google Scholar] [CrossRef] [PubMed]
- Hamed, M.I.; Darwish, K.M.; Soltane, R.; Chrouda, A.; Mostafa, A.; Shama, N.M.A.; Elhady, S.S.; Abulkhair, H.S.; Khodir, A.E.; Elmaaty, A.A. β-Blockers bearing hydroxyethylamine and hydroxyethylene as potential SARS-CoV-2 Mpro inhibitors: Rational based design, in silico, in vitro, and SAR studies for lead optimization. Rsc. Adv. 2021, 11, 35536–35558. [Google Scholar] [CrossRef]
- Huang, Y.-H.; Yeh, C.-T.; Hsu, C.-W.; Lin, Y.-H. Reduction of ACE2 Serum Concentrations by Telbivudine in Chronic Hepatitis B Patients. Curr. Mol. Med. 2022, 23, 420–424. [Google Scholar] [CrossRef]
- Hussein, R.K.; Khouqeer, G.; Alkaoud, A.M.; El-Khayatt, A.M. Probing the Action of Screened Anticancer Triazole–Tetrazole Derivatives against COVID-19 Using Molecular Docking and DFT Investigations. Nat. Prod. Commun. 2022, 17, 1934578X221093915. [Google Scholar] [CrossRef]
- Baig, A.; Srinivasan, H. SARS-CoV-2 Inhibitors from Nigella Sativa. Appl. Biochem. Biotechnol. 2022, 194, 1051–1090. [Google Scholar] [CrossRef]
- Kashyap, P.; Bhardwaj, V.K.; Chauhan, M.; Chauhan, V.; Kumar, A.; Purohit, R.; Kumar, A.; Kumar, S. A ricin-based peptide BRIP from Hordeum vulgare inhibits Mpro of SARS-CoV-2. Sci. Rep. 2022, 12, 12802. [Google Scholar] [CrossRef]
- Mercorelli, B.; Desantis, J.; Celegato, M.; Bazzacco, A.; Siragusa, L.; Benedetti, P.; Eleuteri, M.; Croci, F.; Cruciani, G.; Goracci, L. Discovery of novel SARS-CoV-2 inhibitors targeting the main protease Mpro by virtual screenings and hit optimization. Antivir. Res. 2022, 204, 105350. [Google Scholar] [CrossRef]
- Jatczak, M.; Muylaert, K.; De Coen, L.M.; Keemink, J.; Wuy, B.; Augusti-Jins, P.; Stevens, C.V. Straightforward entry to pyrido [2, 3-d] pyrimidine-2, 4 (1H, 3H)-diones and their ADME properties. Bioorg. Med. Chem. 2014, 22, 3947–3956. [Google Scholar] [CrossRef]
- Arif, R. Design, synthesize and antiurease activity of novel thiazole derivatives: Machine learning, molecular docking and biological investigation. J. Mol. Struct. 2020, 1208, 127905. [Google Scholar] [CrossRef]
- Yousef, T.A.; Khairy, M. Synthesis, Characterization, Optical, DFT, TD DFT Studies and in Silico ADME Predictions of Thiosemicarbazone Ligand and its Au(III) Complex Orient. J. Chem. 2022, 38, 537–546. [Google Scholar] [CrossRef]
- Parvarinezhad, S.; Salehi, M. Synthesis, characterization, crystal structures, Hirshfeld surface analysis and DFT computational studies of new Schiff Bases derived from Phenylhydrazine. J. Mol. Struct. 2020, 1222, 128780. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 1988, 38, 3098–3100. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hehre, W.J.; Ditchfield, R.; Pople, J.A. Self—Consistent Molecular Orbital Methods. XII. Further Extensions of Gaussian—Type Basis Sets for Use in Molecular Orbital Studies of Organic Molecules. J. Chem. Phys. 1972, 56, 2257–2261. [Google Scholar] [CrossRef]
- Rassolov, V.A.; Pople, J.A.; Ratner, M.A.; Windus, T.L. 6-31G* basis set for atoms K through Zn. J. Chem. Phys. 1998, 109, 1223–1229. [Google Scholar] [CrossRef]
- Hay, P.J.; Wadt, W.R. Ab initio effective core potentials for molecular calculations. Potentials for the transition metal atoms Sc to Hg. J. Chem. Phys. 1985, 82, 270–283. [Google Scholar] [CrossRef]
- Wang, J.; Wang, W.; Kollman, P.A.; Case, D.A. Automatic atom type and bond type perception in molecular mechanical calculations. J. Mol. Graph. Model. 2006, 25, 247–260. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elzupir, A.O. Inhibition of SARS-CoV-2 main protease 3CLpro by means of α-ketoamide and pyridone-containing pharmaceuticals using in silico molecular docking. J. Mol. Struct. 2020, 1222, 128878. [Google Scholar] [CrossRef] [PubMed]
- Al-Janabi, A.S.; Elzupir, A.O.; Yousef, T.A. Synthesis, anti-bacterial evaluation, DFT study and molecular docking as a potential 3-chymotrypsin-like protease (3CLpro) of SARS-CoV-2 inhibitors of a novel Schiff bases. J. Mol. Struct. 2021, 1228, 129454. [Google Scholar] [CrossRef] [PubMed]
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open Babel: An open chemical toolbox. J. Cheminform. 2011, 3, 33. [Google Scholar] [CrossRef] [Green Version]
- Shapovalov, M.V.; Dunbrack, R.L., Jr. A smoothed backbone-dependent rotamer library for proteins derived from adaptive kernel density estimates and regressions. Structure 2011, 19, 844–858. [Google Scholar] [CrossRef] [Green Version]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Seq. | Color | % Yield | M.p (°C) | Cond. | % CHN Analysis Found (Calc.) | ||
|---|---|---|---|---|---|---|---|
| C | H | N | |||||
| Cmpy | Off-White | 87 | 178–181 | -- | 63.55 | 4.92 | 17.10 |
| (63.67) | (5.11) | (17.34) | |||||
| 1 | Green | 91 | 230d | 7.9 | 41.60 | 3.22 | 11.20 |
| (41.52) | (3.39) | (11.42) | |||||
| 2 | Brown | 83 | 231–233 | 11.2 | 36.91 | 2.86 | 9.93 |
| (36.78) | (3.07) | (10.11) | |||||
| 3 | Dark yellow | 79 | 189–191 | 10.8 | 30.51 | 2.36 | 8.21 |
| (30.48) | (2.52) | (8.36) | |||||
| 4 | White | 81 | 243–245 | 3.6 | 40.88 | 3.17 | 11.00 |
| (41.03) | (3.39) | (11.23) | |||||
| 5 | Off-White | 87 | 262d | 9.4 | 30.19 | 2.34 | 8.12 |
| (30.43) | (2.27) | (8.29) | |||||
| Band assignments | L | Ni | Pd | Pt | Zn | Hg | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Exp. | Calc. | Exp. | Calc. | Exp. | Calc. | Exp. | Calc. | Exp. | Calc. | Exp. | Calc. | |
| N-H | 3240 | 3219 | 3228 | 3233 | 3134 | 3203 | 3142 | 3157 | 3162 | 3157 | 3136 | 3127 |
| C-H aromatic | 3082 | 3084 | 3055 | 3052 | 2997 | 3041 | 3051 | 3052 | 3068 | 3052 | 2999 | 3051 |
| C-H aliphatic | 2912 | 3023 | 2941 | 2967 | 2918 | 3021 | 2928 | 2945 | 2987 | 2967 | 2958 | 2978 |
| C=N pyridyl ring | 1689 | 1656 | 1622 | 1620 | 1595 | 1590 | 1616 | 1616 | 1622 | 1628 | 1595 | 1599 |
| C=N azomethine | 1606 | 1615 | 1531 | 1528 | 1548 | 1547 | 1531 | 1530 | 1531 | 1528 | 1550 | 1573 |
| C=C | 1525 | 1516 | 1481 | 1476 | 1479 | 1490 | 1485 | 1479 | 1433 | 1442 | 1479 | 1471 |
| CH3 bending (rock) | 1398 | 1381 | 1385 | 1398 | 1384 | 1365 | 1390 | 1389 | 1388 | 1398 | 1384 | 1399 |
| N-N | 1361 | 1339 | 1340 | 1341 | 1334 | 1334 | 1338 | 1338 | 1338 | 1336 | 1344 | 1362 |
| C-N | 1278 | 1278 | 1246 | 1232 | 1247 | 1240 | 1246 | 1238 | 1249 | 1226 | 1247 | 1272 |
| CH3 wag. | 1126 | 1118 | 1122 | 1138 | 1120 | 1140 | 1118 | 1090 | 1116 | 1089 | 1153 | 1158 |
| In plane =C-H bending | 947 | 967 | 929 | 934 | 918 | 925 | 923 | 922 | 925 | 921 | 925 | 922 |
| C-Cl | 817 | 834 | 822 | 806 | 842 | 815 | 842 | 846 | 837 | 856 | 840 | 853 |
| oop C-H bending | 756 | 749 | 736 | 737 | 746 | 751 | 748 | 732 | 754 | 739 | 759 | 738 |
| M-N | - | 509 | 497 | 507 | 523 | 511 | 533 | 489 | 474 | 447 | 447 | |
| Comps. | δH (ppm) and Assignments | |||
|---|---|---|---|---|
| NH | CH=N | Aromatic Protons | CH3 | |
| Cmpy | 11.25 (s, 1H) | 7.80 (s, 1H) | 8.62(d, 1H, J 8.00Hz, H5), 7.92(d, 1H, J 8.00Hz, H3), 7.67(s, 1H, H8), 7.55(dd, 1H, J 8.00Hz, H4), 7.25–7.33(m, 3H, H2,12,11), | 2.38 (s, 3H) |
| 1 | 11.05 (s, 1H) | 7.83 (s, 1H) | 8.62(d, 1H, J 8.00Hz, H5), 7.92(d, 1H, J 8.00Hz, H3), 7.67(s, 1H, H8), 7.53(dd, 1H, J 8.00Hz, H4), 7.25–7.367.25–7.33(m, 3H, H2,12,11), | 2.37 (s, 3H) |
| 2 | 10.89 (s, 1H) | 7.80 (s, 1H) | 8.61(dd, 1H, J 8.00Hz, H5), 7.85(d, 1H, J 7.80Hz, H3), 7.63(s, 1H, H8), 7.56(t, 1H, J 8.00Hz, H4) 7.30–7.33(m, 3H, H2,11,12) | 2.29 (s, 3H) |
| 3 | 11.04 (s, 1H) | 7.85 (s, 1H) | 8.63(dd, 1H, J 8.00Hz, H5), 8.01(dt, 1H, J 7.80Hz, H3), 7.77(s, 1H, H8), 7.69(d, J 8.00Hz, 1H, H2), 7.63(d, 1H, J 8.00Hz, H11) 7.47(t, 1H, H4), | 2.19 (s, 3H) |
| 4 | 11.09 (s, 1H) | Overlap with aromatic protons | 8.67(d, 1H, J 7.60Hz, H5), 7.62–7.92(m, 4H, H2,8,12, CH=N), 7.51(d, J 8.00Hz, 1H, H3), 7.36(d, 1H, J 7.60Hz, H11) 7.30(t, 1H, J 7.80Hz, H4) | 2.10 (s, 3H) |
| 5 | 10.96 (s, 1H) | Overlap with aromatic protons | 8.67(d, 1H, J 8.00Hz, H5), 7.48–7.92(m, 5H, H2,3,8,12,CH=N), 7.37(dd, 1H, J 8.00Hz, H11) 7.30(t, 1H, J 8.00Hz, H4) | 2.18 (s, 3H) |
| Comps. | δC (ppm) | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| (CH=N) | C1 | C2 | C3 | C4 | C5 | C7 | C8 | C9 | C10 | C11 | C12 | CH3 | |
| Cmpy | 162.12 | 132.41 | 129.83 | 129.39 | 129.30 | 155.75 | 140.92 | 122.36 | 131.01 | 131.04 | 129.34 | 120.21 | 22.02 |
| 1 | 160.04 | 132.41 | 128.83 | 129.39 | 129.27 | 152.66 | 140.39 | 121.85 | 129.48 | 131.07 | 129.30 | 119.51 | 21.50 |
| 2 | 159.30 | 132.85 | 129.30 | 129.99 | 129.46 | 152.85 | 141.04 | 122.62 | 130.85 | 131.48 | 129.69 | 120.71 | 22.80 |
| 5 | 161.14 | 133.48 | 127.67 | 129.87 | 128.07 | 153.45 | 142.45 | 122.91 | 130.77 | 132.45 | 129.23 | 121.78 | 22.78 |
| Seq. | S. aureus | P. aeruginosa | ||
|---|---|---|---|---|
| DIZ (mm) ± SD | A.I. (%) | DIZ (mm) ± SD | A.I. (%) | |
| Cmpy | 14 ± 1.04 | 48 | 15 ± 0.45 | 48 |
| 1 | 22 ± 0.91 | 76 | 25 ± 0.49 | 81 |
| 2 | 19 ± 0.78 | 66 | 22 ± 0.61 | 71 |
| 3 | 19 ± 0.92 | 66 | 20 ± 1.02 | 65 |
| 4 | 25 ± 1.10 | 86 | 28 ± 1.10 | 90 |
| 5 | 18 ± 0.78 | 62 | 22 ± 0.71 | 71 |
| Amoxicillin | 29 ± 1.04 | 100 | 31 ± 0.61 | 100 |
| Parameter | Cmpy | Complexes | ||||
|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | ||
| Electronic Energy | −1127.23 | −881.01 | −832.47 | −830.85 | −777.32 | −754.42 |
| Thermal Energy | 150.19 | 158.86 | 168.91 | 158.38 | 158.49 | 158.16 |
| Total Dipole Moment | 4.98 | 11.13 | 13.76 | 11.13 | 10.92 | 10.23 |
| Polarizability (α) | 211.62 | 246.87 | 230.33 | 270.78 | 228.90 | 244.68 |
| Heat Capacity (Cv) | 57.72 | 71.12 | 65.63 | 69.52 | 72.47 | 72.93 |
| Entropy (S) | 127.28 | 148.33 | 141.21 | 144.42 | 156.37 | 161.81 |
| Comp. | EH / eV | EL / eV | (EL-EH) /Ev | χ / eV | μ / eV | η / eV | S / eV−1 | ω / eV | σ / eV−1 |
|---|---|---|---|---|---|---|---|---|---|
| Cmpy | −5.58 | −1.87 | 3.71 | 3.73 | −3.73 | 1.86 | 0.54 | 3.74 | 0.93 |
| Ni complex | −6.40 | −3.14 | 3.26 | 4.77 | −4.77 | 1.63 | 0.61 | 6.98 | 0.82 |
| Pd complex | −3.16 | −0.72 | 2.44 | 1.94 | −1.94 | 1.22 | 0.82 | 1.54 | 0.61 |
| Pt complex | −6.35 | −3.16 | 3.19 | 4.76 | −4.76 | 1.59 | 0.79 | 7.09 | 0.63 |
| Zn complex | −6.67 | −3.23 | 3.44 | 4.95 | −4.95 | 1.72 | 0.58 | 7.12 | 0.86 |
| Hg complex | −6.48 | −2.86 | 3.62 | 4.67 | −4.67 | 1.81 | 0.55 | 6.02 | 0.91 |
| Pharmaceutical Name | Binding Percentage a | Score (kcal/mol) | RMSD (L–H) b | Hydrogen Bond (Number of Bonds/Number of Conformations), (Distance Range Å) | Van der Waals (Number of Bonds/Number of Conformations), (Distance Range Å) |
|---|---|---|---|---|---|
| Sinefungin | 100 | –6.6 to –7.5 | 0.00–7.76 | LYS 6844 (2/2), (2.529–2.709) LEU 6898 (1/1), (2.231) TYR 6930 (3/3), (1.967–2.539) LYS 6968 (1/1), (2.286) ASN 6996 (2/1), (2.198–2.255) SER 6999 (1/1), (2.383) ASP 6912 (1/1), (2.489) ASP 6897 (2/2), (2.039–2.250) GLY 6869 (1/1), (2.366) ASP 6928 (1/1), (2.589) GLY 6871 (3/3), (1.933–2.169) ASP 6897 (2/2), (2.039–2.250) GLY 6869 (1/1), (2.366) ASP 6928 (1/1), (2.589) GLU 7001 (1/1), (2.281) ASP 6873 (1/1), (2.355) | ASP 6928 (34/9), (1.699–3.963), ASP 6897 (35/7), (2.039–3.728), ASP 6912 (2/1), (2.489–3.150), |
| Ligand | 78 | –5.8 to –6.7 | 0.00–8.344 | LYS 6844 (1/1), (2.304) TYR 6930 (1/1), (2.486) LYS 6935 (1/1), (2.597) LYS 7051 (1/1), (2.210) ASN 6841 (1/1), (2.620) ASP 6928 (1/1), (2.355) SER 6999 (1/1), (1.878) GLY 6871 (1/1), (1.835) | ASP 6928 (14/4), (2.355–3.490), ASP 6897 (18/7), (3.115–4.008), ASP 6912 (3/3), (3.417–3.685), |
| Ni complex | 100 | –6.0 to –7.1 | 0.00–8.08 | ASP 6928 (1/1), (2.454) GLY 6871 (2/2), (1.963–2.294) ASN 6841 (1/1), (2.486) | ASP 6928 (29/8), (2.454–4.058), ASP 6897 (6/4), (2.263–3.500), ASP 6912 (5/3), (3.685) |
| Zn complex | 100 | –6.2 to –7.2 | 0.00–8.04 | ASP 6897 (2/2), (2.445–2.478) ASP 6928 (1/1), (2.397) GLY 6871 (1/1), 2.198 ASN 6841 (1/1), (2.294) | ASP 6928 (25/9), (2.397–3.928), ASP 6897 (15/6), (2.445–3.638), ASP 6912 (6/3), (2.210–3.719) |
| Pharmaceutical Name | Binding Percentage a | Score ± SD (kcal/mol) b | RMSD (L–H) | Hydrogen Bond (Number of Bonds/Number of Conformations), (Distance Range Å) | Van der Waals (Number of Bonds/Number of Conformations), (Distance Range Å) |
|---|---|---|---|---|---|
| Ni Complex | 22 | −5.9 to –6.0 | 29.46–32.52 | - | GLU 166 (11/1), (2.022–3.722) CYS 145 (7/2), (2.956–3.863) HIS 41 (16/2), (1.886–3.949) |
| Zn Complex | 22 | −6.2 to −7.2 | 0.00–4.22 | LEU 287 (1/1), (2.572) LEU 75 (1/1), (2.023) | GLU 166 (12/2), (2.653–3.789) CYS 145 (5/2), (2.860–3.881) HIS 41 (8/2), (2.079–3.784) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Al-Janabi, A.S.M.; Elzupir, A.O.; Abou-Krisha, M.M.; Yousef, T.A. New Dual Inhibitors of SARS-CoV-2 Based on Metal Complexes with Schiff-Base 4-Chloro-3-Methyl Phenyl Hydrazine: Synthesis, DFT, Antibacterial Properties and Molecular Docking Studies. Inorganics 2023, 11, 63. https://doi.org/10.3390/inorganics11020063
Al-Janabi ASM, Elzupir AO, Abou-Krisha MM, Yousef TA. New Dual Inhibitors of SARS-CoV-2 Based on Metal Complexes with Schiff-Base 4-Chloro-3-Methyl Phenyl Hydrazine: Synthesis, DFT, Antibacterial Properties and Molecular Docking Studies. Inorganics. 2023; 11(2):63. https://doi.org/10.3390/inorganics11020063
Chicago/Turabian StyleAl-Janabi, Ahmed S. M., Amin O. Elzupir, Mortaga M. Abou-Krisha, and Tarek A. Yousef. 2023. "New Dual Inhibitors of SARS-CoV-2 Based on Metal Complexes with Schiff-Base 4-Chloro-3-Methyl Phenyl Hydrazine: Synthesis, DFT, Antibacterial Properties and Molecular Docking Studies" Inorganics 11, no. 2: 63. https://doi.org/10.3390/inorganics11020063