


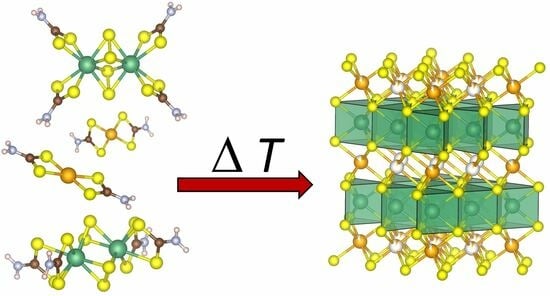
Nb2S4(CS2NH2)4—A New Precursor for NbS2 and Its Transition Metal Inserted Derivatives

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
2.1. Synthesis
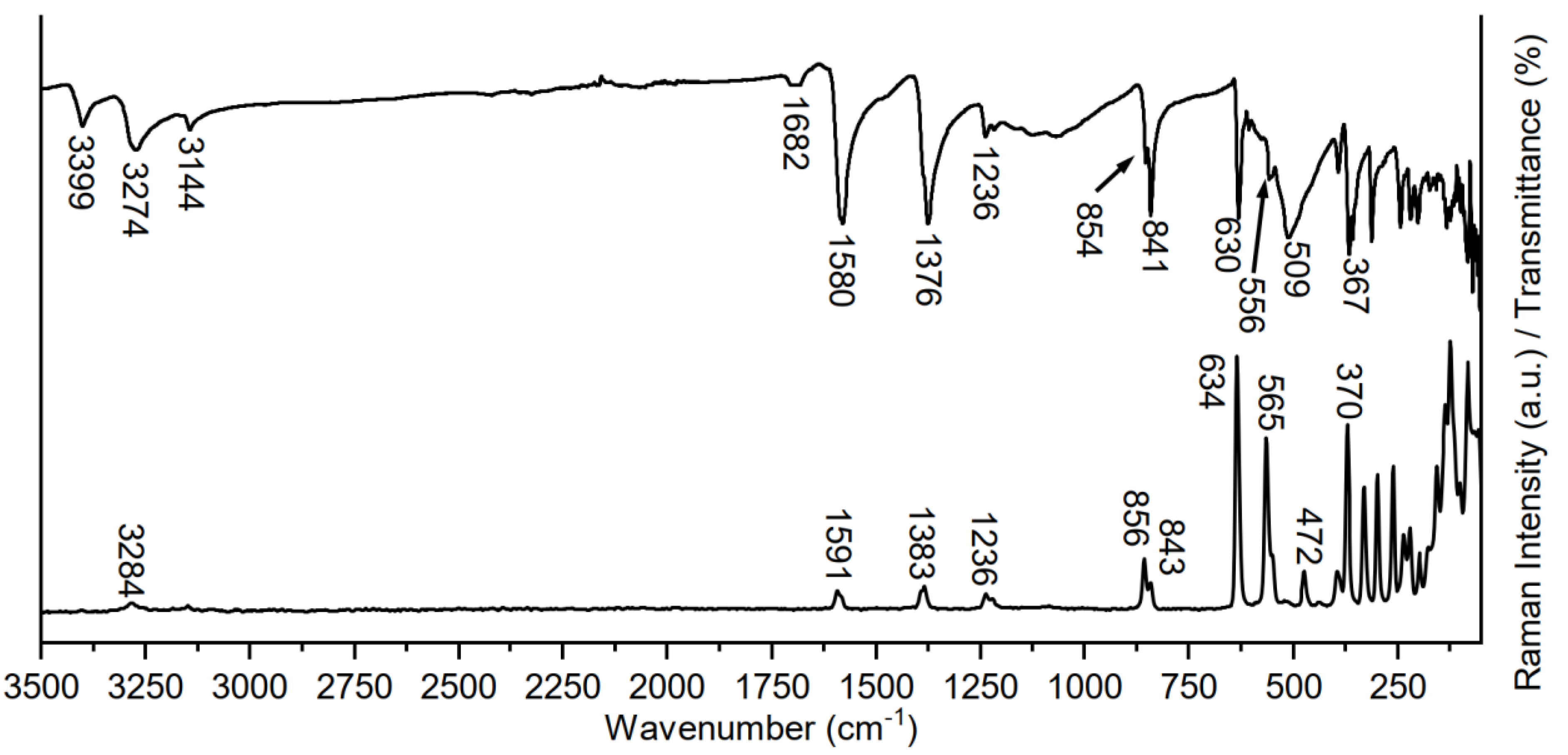
2.2. Vibrational Spectroscopy
2.3. Crystal Structures
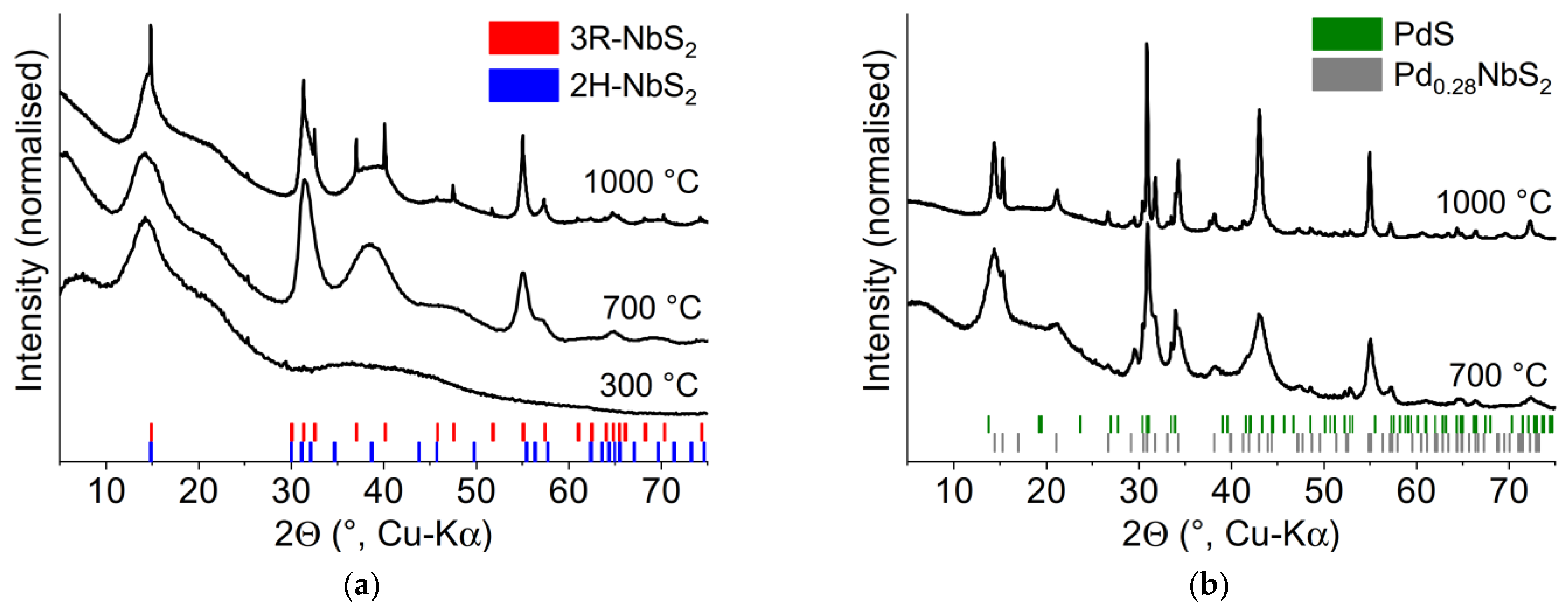
2.4. Thermal Decomposition
3. Materials and Methods
3.1. Preparations
3.2. X-ray Powder Diffraction
3.3. Single Crystal X-ray Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Wilson, J.A.; Yoffe, A.D. The Transition Metal Dichalcogenides Discussion and Interpretation of the Observed Optical, Electrical and Structural Properties. Adv. Phys. 1969, 18, 193–335. [Google Scholar] [CrossRef]
- Katzke, H.; Tolédano, P.; Depmeier, W. Phase Transitions between Polytypes and Intralayer Superstructures in Transition Metal Dichalcogenides. Phys. Rev. B 2004, 69, 134111. [Google Scholar] [CrossRef]
- Ali, M.N.; Xiong, J.; Flynn, S.; Tao, J.; Gibson, Q.D.; Schoop, L.M.; Liang, T.; Haldolaarachchige, N.; Hirschberger, M.; Ong, N.P.; et al. Large, Non-Saturating Magnetoresistance in WTe2. Nature 2014, 514, 205–208. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Li, H.; Dai, Y.M.; Miao, H.; Shi, Y.G.; Ding, H.; Taylor, A.J.; Yarotski, D.A.; Prasankumar, R.P.; Thompson, J.D. Hall Effect in the Extremely Large Magnetoresistance Semimetal WTe2. Appl. Phys. Lett. 2015, 107, 182411. [Google Scholar] [CrossRef]
- Chen, F.C.; Lv, H.Y.; Luo, X.; Lu, W.J.; Pei, Q.L.; Lin, G.T.; Han, Y.Y.; Zhu, X.B.; Song, W.H.; Sun, Y.P. Extremely Large Magnetoresistance in the Type-II Weyl Semimetal MoTe2. Phys. Rev. B 2016, 94, 235154. [Google Scholar] [CrossRef]
- Thirupathaiah, S.; Jha, R.; Pal, B.; Matias, J.S.; Das, P.K.; Sivakumar, P.K.; Vobornik, I.; Plumb, N.C.; Shi, M.; Ribeiro, R.A.; et al. MoTe2: An Uncompensated Semimetal with Extremely Large Magnetoresistance. Phys. Rev. B 2017, 95, 241105. [Google Scholar] [CrossRef]
- Mangelsen, S.; Naumov, P.G.; Barkalov, O.I.; Medvedev, S.A.; Schnelle, W.; Bobnar, M.; Mankovsky, S.; Polesya, S.; Näther, C.; Ebert, H.; et al. Large Nonsaturating Magnetoresistance and Pressure-Induced Phase Transition in the Layered Semimetal HfTe2. Phys. Rev. B 2017, 96, 205148. [Google Scholar] [CrossRef]
- Mangelsen, S.; Bensch, W. HfTe2: Enhancing Magnetoresistance Properties by Improvement of the Crystal Growth Method. Inorg. Chem. 2020, 59, 1117–1124. [Google Scholar] [CrossRef]
- Pletikosić, I.; Ali, M.N.; Fedorov, A.V.; Cava, R.J.; Valla, T. Electronic Structure Basis for the Extraordinary Magnetoresistance in WTe2. Phys. Rev. Lett. 2014, 113, 216601. [Google Scholar] [CrossRef]
- Wu, Y.; Mou, D.; Jo, N.H.; Sun, K.; Huang, L.; Bud’ko, S.L.; Canfield, P.C.; Kaminski, A. Observation of Fermi Arcs in the Type-II Weyl Semimetal Candidate WTe2. Phys. Rev. B 2016, 94, 121113. [Google Scholar] [CrossRef]
- Xu, C.; Li, B.; Jiao, W.; Zhou, W.; Qian, B.; Sankar, R.; Zhigadlo, N.D.; Qi, Y.; Qian, D.; Chou, F.-C.; et al. Topological Type-II Dirac Fermions Approaching the Fermi Level in a Transition Metal Dichalcogenide NiTe2. Chem. Mater. 2018, 30, 4823–4830. [Google Scholar] [CrossRef]
- Sun, Y.; Wu, S.-C.; Ali, M.N.; Felser, C.; Yan, B. Prediction of Weyl Semimetal in Orthorhombic MoTe2. Phys. Rev. B 2015, 92, 161107. [Google Scholar] [CrossRef]
- Deng, K.; Wan, G.; Deng, P.; Zhang, K.; Ding, S.; Wang, E.; Yan, M.; Huang, H.; Zhang, H.; Xu, Z.; et al. Experimental Observation of Topological Fermi Arcs in Type-II Weyl Semimetal MoTe2. Nat. Phys. 2016, 12, 1105–1110. [Google Scholar] [CrossRef]
- Witteveen, C.; Górnicka, K.; Chang, J.; Månsson, M.; Klimczuk, T.; Rohr, F.O. von Polytypism and Superconductivity in the NbS2 System. Dalton Trans. 2021, 50, 3216–3223. [Google Scholar] [CrossRef] [PubMed]
- Castro Neto, A.H. Charge Density Wave, Superconductivity, and Anomalous Metallic Behavior in 2D Transition Metal Dichalcogenides. Phys. Rev. Lett. 2001, 86, 4382–4385. [Google Scholar] [CrossRef] [PubMed]
- Dutta, U.; Malavi, P.S.; Sahoo, S.; Joseph, B.; Karmakar, S. Pressure-Induced Superconductivity in Semimetallic 1T-TiTe2 and Its Persistence upon Decompression. Phys. Rev. B 2018, 97, 060503. [Google Scholar] [CrossRef]
- Klemm, R.A. Pristine and Intercalated Transition Metal Dichalcogenide Superconductors. Phys. C 2015, 514, 86–94. [Google Scholar] [CrossRef]
- Marseglia, E.A. Transition Metal Dichalcogenides and Their Intercalates. Int. Rev. Phys. Chem. 1983, 3, 177–216. [Google Scholar] [CrossRef]
- Lerf, A. Storylines in Intercalation Chemistry. Dalton Trans. 2014, 43, 10276–10291. [Google Scholar] [CrossRef]
- Parkin, S.S.P.; Friend, R.H. 3d Transition-Metal Intercalates of the Niobium and Tantalum Dichalcogenides. I. Magnetic Properties. Philos. Mag. B 1980, 41, 65–93. [Google Scholar] [CrossRef]
- Inoue, M.; Hughes, H.P.; Yoffe, A.D. The Electronic and Magnetic Properties of the 3d Transition Metal Intercalates of TiS2. Adv. Phys. 1989, 38, 565–604. [Google Scholar] [CrossRef]
- Koyano, M.; Horisaka, S.; Negishi, H.; Sasaki, M.; Inoue, M.; Suzuki, N.; Motizuki, K. Magnetic Scattering of Conduction Carriers in 3d Transition-Metal Intercalates of MxTiS2 (M = Mn, Fe, Co, and Ni). J. Low Temp. Phys. 1990, 78, 141–154. [Google Scholar] [CrossRef]
- Voorhoeve, J.M.; van den Berg, N.; Robbins, M. Intercalation of the Niobium-Diselenide Layer Structure by First-Row Transition Metals. J. Solid State Chem. 1970, 1, 134–137. [Google Scholar] [CrossRef]
- Yacobi, B.G.; Boswell, F.W.; Corbett, J.M. The Fundamental Absorption Edge in NixZrS2. Mater. Res. Bull. 1979, 14, 1033–1038. [Google Scholar] [CrossRef]
- Buhannic, M.; Danot, M. Iron Migration in the Van Der Waals Gap of Zirconium Disulfide: Time Evolution of the FexZrS2 Compounds. Solid State Commun. 1990, 73, 739–742. [Google Scholar] [CrossRef]
- Parkin, S.S.P.; Friend, R.H. 3d Transition-Metal Intercalates of the Niobium and Tantalum Dichalcogenides. II. Transport Properties. Philos. Mag. B 1980, 41, 95–112. [Google Scholar] [CrossRef]
- Chen, C.-W.; Chikara, S.; Zapf, V.S.; Morosan, E. Correlations of Crystallographic Defects and Anisotropy with Magnetotransport Properties in FexTaS2 Single Crystals 0.23 ≤ x ≤ 0.35. Phys. Rev. B 2016, 94, 054406. [Google Scholar] [CrossRef]
- Inoshita, T.; Hirayama, M.; Hamada, N.; Hosono, H.; Murakami, S. Topological Semimetal Phases Manifested in Transition Metal Dichalcogenides Intercalated with 3d Metals. Phys. Rev. B 2019, 100, 121112. [Google Scholar] [CrossRef]
- Maniv, E.; Nair, N.L.; Haley, S.C.; Doyle, S.; John, C.; Cabrini, S.; Maniv, A.; Ramakrishna, S.K.; Tang, Y.-L.; Ercius, P.; et al. Antiferromagnetic Switching Driven by the Collective Dynamics of a Coexisting Spin Glass. Sci. Adv. 2021, 7, eabd8452. [Google Scholar] [CrossRef]
- Le Blanc-Soreau, A.; Rouxel, J.; Gardette, M.-F.; Gorochov, O. Proprietes electriques et magnetiques de Mn0,25NbS2 et Mn0,33NbS2. Mater. Res. Bull. 1976, 11, 1061–1071. [Google Scholar] [CrossRef]
- Yamasaki, Y.; Moriya, R.; Arai, M.; Masubuchi, S.; Pyon, S.; Tamegai, T.; Ueno, K.; Machida, T. Exfoliation and van Der Waals Heterostructure Assembly of Intercalated Ferromagnet Cr1/3TaS2. 2D Mater. 2017, 4, 041007. [Google Scholar] [CrossRef]
- Arai, M.; Moriya, R.; Yabuki, N.; Masubuchi, S.; Ueno, K.; Machida, T. Construction of van Der Waals Magnetic Tunnel Junction Using Ferromagnetic Layered Dichalcogenide. Appl. Phys. Lett. 2015, 107, 103107. [Google Scholar] [CrossRef]
- Danz, T.; Liu, Q.; Zhu, X.D.; Wang, L.H.; Cheong, S.W.; Radu, I.; Ropers, C.; Tobey, R.I. Structural and Magnetic Characterization of Large Area, Free-Standing Thin Films of Magnetic Ion Intercalated Dichalcogenides Mn0.25TaS2 and Fe0.25TaS2. J. Phys. Condens. Matter 2016, 28, 356002. [Google Scholar] [CrossRef] [PubMed]
- Ellermeier, J.; Näther, C.; Bensch, W. Tris(Ethylenediamine-N,N’)Nickel(II) Tetrathiomolybdate. Acta Crystallogr. Sect. C Cryst. Struct. Commun. 1999, 55, 501–503. [Google Scholar] [CrossRef]
- Ellermeier, J.; Stähler, R.; Bensch, W. Two New [Ni(Tren)2]2+ Complexes: [Ni(Tren)2]Cl2 and [Ni(Tren)2]WS4. Acta Crystallogr. Sect. C Cryst. Struct. Commun. 2002, 58, m70–m73. [Google Scholar] [CrossRef]
- Ellermeier, J.; Bensch, W. Solvothermal Syntheses, Crystal Structures and Properties of Thiomolybdates with Complex Transition Metal Cations. Z. Naturforsch. B J. Chem. Sci. 2001, 56, 611–619. [Google Scholar] [CrossRef]
- Sokolov, M.; Virovets, A.; Nadolinnyi, V.; Hegetschweiler, K.; Fedin, V.; Podberezskaya, N.; Fedorov, V. Nb2S24+ Complexes with 1,1-Dithioacid Ligands. Inorg. Chem. 1994, 33, 3503–3509. [Google Scholar] [CrossRef]
- Sengupta, S.K.; Kumar, S. Thermal Studies on Metal Dithiocarbamato Complexes. A Review. Thermochim. Acta 1984, 72, 349–361. [Google Scholar] [CrossRef]
- Sharma, A.K. Thermal Behaviour of Metal-Dithiocarbamates. Thermochim. Acta 1986, 104, 339–372. [Google Scholar] [CrossRef]
- Poisot, M.; Bensch, W.; Fuentes, S.; Alonso, G. Decomposition of Tetra-Alkylammonium Thiomolybdates Characterised by Thermoanalysis and Mass Spectrometry. Thermochim. Acta 2006, 444, 35–45. [Google Scholar] [CrossRef]
- Poisot, M.; Bensch, W. Decomposition of Tetraalkylammonium Thiotungstates Characterized by Thermoanalysis, Mass Spectrometry, X-Ray Diffractometry and Scanning Electron Microscopy. Thermochim. Acta 2007, 453, 42–51. [Google Scholar] [CrossRef]
- Sokolov, M.N.; Rogachev, A.V.; Abramov, P.A.; Fedin, V.P. Thioxanthate Complexes of {Nb2S4}4+. Polyhedron 2015, 85, 727–731. [Google Scholar] [CrossRef]
- Raston, C.L.; White, A.H.; Willis, A.C. Crystal Structure of Tris(Dithiocarbamato)Cobalt(III). J. Chem. Soc. Dalton Trans. 1975, 2429–2432. [Google Scholar] [CrossRef]
- Gasparri, G.F.; Nardelli, M.; Villa, A. The Crystal and Molecular Structure of Nickel Bis(Dithiocarbamate). Acta Cryst. 1967, 23, 384–391. [Google Scholar] [CrossRef]
- Nakamoto, K.; Fujita, J.; Condrate, R.A.; Morimoto, Y. Infrared Spectra of Metal Chelate Compounds. IX. A Normal Coordinate Analysis of Dithiocarbamato Complexes. J. Chem. Phys. 1963, 39, 423–427. [Google Scholar] [CrossRef]
- Rijnsdorp, J.; Haas, C. Fine Structure in the Absorption Edge Spectrum of NbS2Y2 (Y = Cl, Br, I). J. Phys. Chem. Solids 1980, 41, 375–384. [Google Scholar] [CrossRef]
- Kato, K.; Tamura, S. Die Kristallstruktur von T-Nb2O5. Acta Crystallogr. Sect. B Struct. Crystallogr. Cryst. Chem. 1975, 31, 673–677. [Google Scholar] [CrossRef]
- Jellinek, F.; Brauer, G.; Müller, H. Molybdenum and Niobium Sulphides. Nature 1960, 185, 376–377. [Google Scholar] [CrossRef]
- Katzke, H. Stacking Disorder in 2H-NbS2 and Its Intercalation Compounds Kx(H2O)yNbS2 I. Description and Model Calculations of Stacking Faults in the Host Lattice NbS2. Z. Kristallogr. Cryst. Mater. 2009, 217, 127–130. [Google Scholar] [CrossRef]
- Fisher, W.G.; Sienko, M.J. Stoichiometry, Structure, and Physical Properties of Niobium Disulfide. Inorg. Chem. 1980, 19, 39–43. [Google Scholar] [CrossRef]
- Huang, C.; Wang, X.; Wang, D.; Zhao, W.; Bu, K.; Xu, J.; Huang, X.; Bi, Q.; Huang, J.; Huang, F. Atomic Pillar Effect in PdxNbS2 To Boost Basal Plane Activity for Stable Hydrogen Evolution. Chem. Mater. 2019, 31, 4726–4731. [Google Scholar] [CrossRef]
- Wiegers, G.A.; Haange, R.J.; van Bolhuis, F. The Crystal Structure of Stage-2 4H-Ag0.22NbS2. Phys. Status Solidi A 1988, 107, 817–824. [Google Scholar] [CrossRef]
- Van der Lee, A.; van Smaalen, S.; Wiegers, G.A.; de Boer, J.L. Order-Disorder Transition in Silver-Intercalated Niobium Disulfide Compounds. I. Structural Determination of Ag0.6NbS2. Phys. Rev. B 1991, 43, 9420–9430. [Google Scholar] [CrossRef] [PubMed]
- Mangelsen, S.; Srinivasan, B.R.; Schürmann, U.; Kienle, L.; Näther, C.; Bensch, W. Nanostructured Tungsten Sulfides: Insights into Precursor Decomposition and the Microstructure Using X-Ray Scattering Methods. Dalton Trans. 2019, 48, 1184–1201. [Google Scholar] [CrossRef] [PubMed]
- Van den Berg, J.M.; Cossee, P. Structural Aspects and Magnetic Behaviour of NbS2 and TaS2 Containing Extra Metal Atoms of the First Transition Series. Inorg. Chim. Acta 1968, 2, 143–148. [Google Scholar] [CrossRef]
- Rijnsdorp, J.; de Lange, G.J.; Wiegers, G.A. Preparation, Structures, and Properties of Niobium Chalcogenide Halides, NbX2Y2 (X = S, Se; Y = Cl, Br, I). J. Solid State Chem. 1979, 30, 365–373. [Google Scholar] [CrossRef]
- Teske, C.L.; Bensch, W. On Crystal Structure Investigations of α- and β-Ammoniumdithiocarbamate NH4CS2NH2 and the Role of Hydrogen Bonding. Z. Anorg. Allg. Chem. 2010, 636, 356–362. [Google Scholar] [CrossRef]
- Schäfer, H.; Beckmann, W. Beiträge Zur Chemie der Elemente Niob und Tantal. LIV. Sulfid- und Selenidhalogenide des Niobs. Z. Anorg. Allg. Chem. 1966, 347, 225–230. [Google Scholar] [CrossRef]
- Schnering, H.G.v.; Beckmann, W. Beiträge zur Chemie der Elemente Niob und Tantal. LV. Die Kristallstruktur von NbS2Cl2. Z. Anorg. Allg. Chem. 1966, 347, 231–239. [Google Scholar] [CrossRef]
- Coelho, A.A. TOPAS and TOPAS-Academic: An Optimization Program Integrating Computer Algebra and Crystallographic Objects Written in C++. J. Appl. Cryst. 2018, 51, 210–218. [Google Scholar] [CrossRef]
- Coelho, A.A. Indexing of Powder Diffraction Patterns by Iterative Use of Singular Value Decomposition. J. Appl. Cryst. 2003, 36, 86–95. [Google Scholar] [CrossRef]
- Coelho, A.A. Whole-Profile Structure Solution from Powder Diffraction Data Using Simulated Annealing. J. Appl. Cryst. 2000, 33, 899–908. [Google Scholar] [CrossRef]
- CrysAlis PRO, version 171.43, Program Package for Crystal Structure Determinations; Rigaku: Wroclow, Poland, 2020.
- X-Area, Version 1.44, Program Package for Crystal Structure Determinations; STOE & Cie GmbH: Darmstadt, Germany, 2002.
- Sheldrick, G.M. SHELXT—Integrated Space-Group and Crystal-Structure Determination. Acta Crystallogr. Sect. A Found. Adv. 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. Crystal Structure Refinement with SHELXL. Acta Crystallogr. Sect. C Struct. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mangelsen, S.; Zimmer, P.; Näther, C.; Bensch, W. Nb2S4(CS2NH2)4—A New Precursor for NbS2 and Its Transition Metal Inserted Derivatives. Inorganics 2023, 11, 478. https://doi.org/10.3390/inorganics11120478
Mangelsen S, Zimmer P, Näther C, Bensch W. Nb2S4(CS2NH2)4—A New Precursor for NbS2 and Its Transition Metal Inserted Derivatives. Inorganics. 2023; 11(12):478. https://doi.org/10.3390/inorganics11120478
Chicago/Turabian StyleMangelsen, Sebastian, Patrick Zimmer, Christian Näther, and Wolfgang Bensch. 2023. "Nb2S4(CS2NH2)4—A New Precursor for NbS2 and Its Transition Metal Inserted Derivatives" Inorganics 11, no. 12: 478. https://doi.org/10.3390/inorganics11120478