
Heterogenous Preparations of Solution-Processable Cobalt Phthalocyanines for Carbon Dioxide Reduction Electrocatalysis

Abstract
:1. Introduction
2. Results
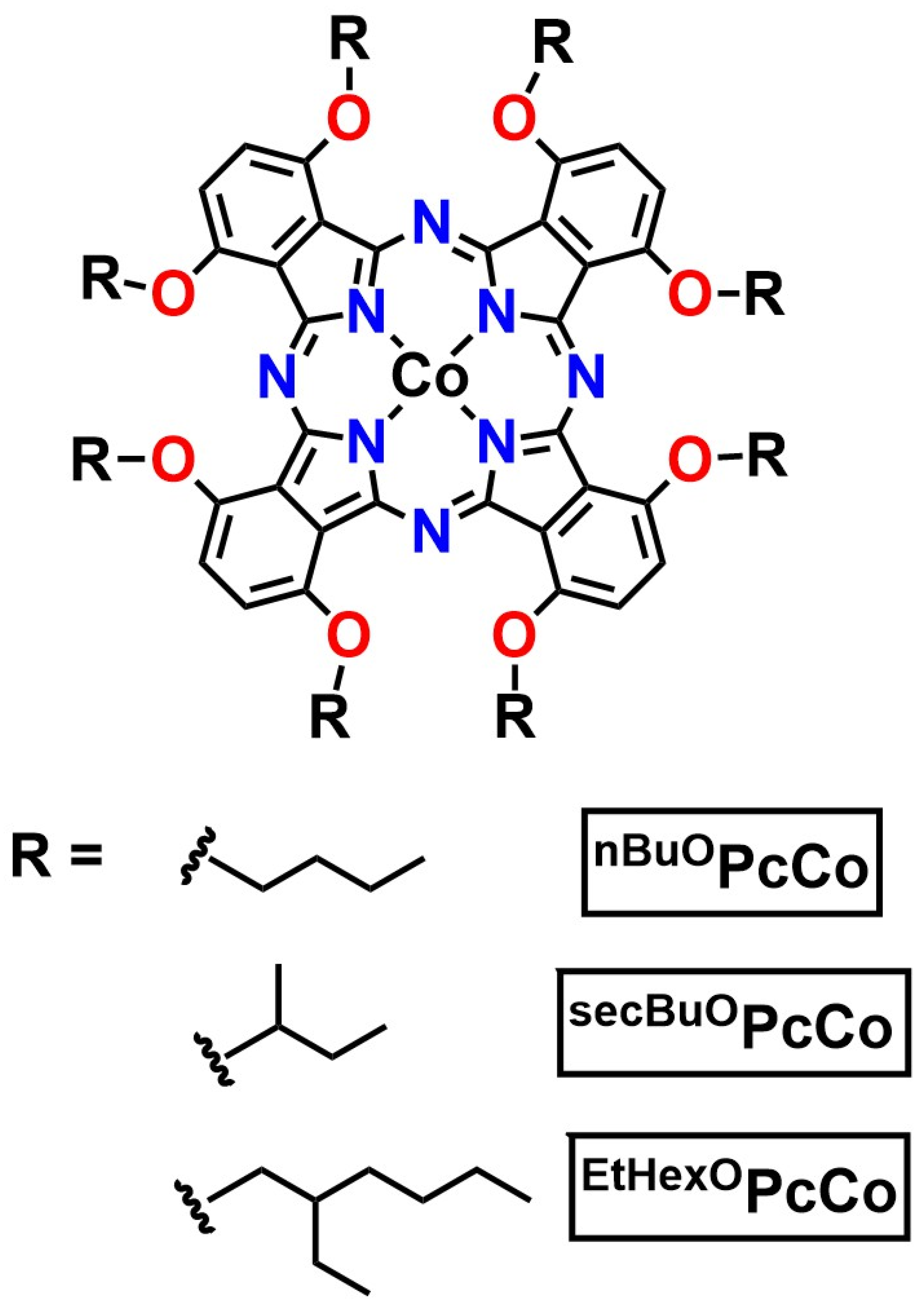
2.1. Design and Synthesis of ROPcCo
2.2. Solution Aggregation of ROPcCo Compounds
2.3. Solution Electrochemistry
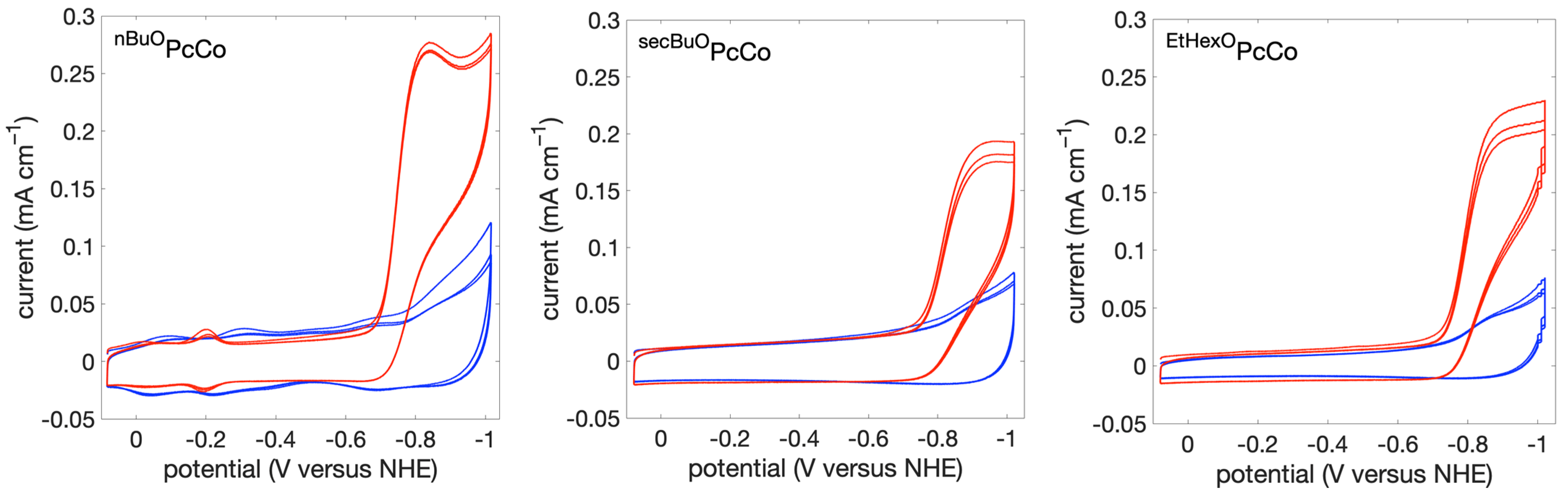
2.4. Heterogeneous Electrochemistry under Argon
2.5. Heterogeneous Electrochemistry under Carbon Dioxide
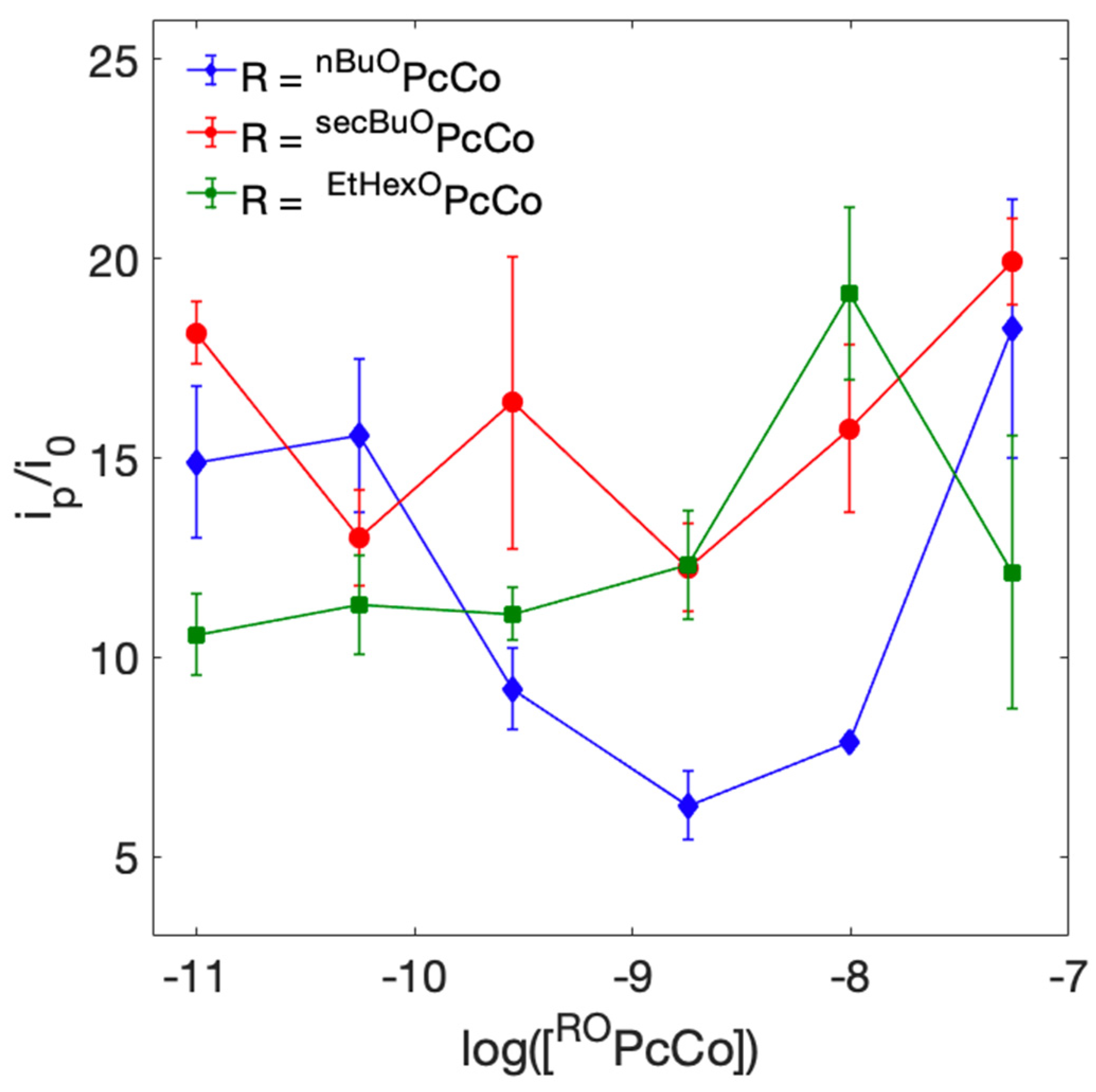
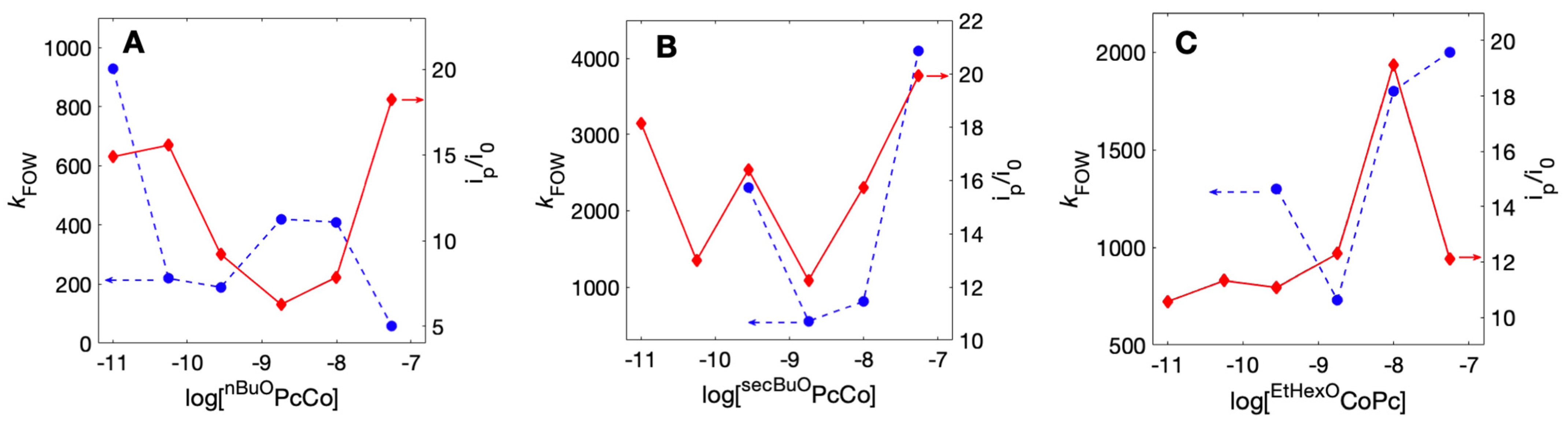
2.6. Quantification of Electroactive ROPcCo
3. Discussion
4. Materials and Methods
4.1. General Materials and Methods
4.2. Synthesis
4.3. Electrochemical Methods
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Whipple, D.T.; Kenis, P.J.A. Prospects of CO2 Utilization via Direct Heterogeneous Electrochemical Reduction. J. Phys. Chem. Lett. 2010, 1, 3451–3458. [Google Scholar] [CrossRef]
- Jouny, M.; Luc, W.; Jiao, F. General Techno-Economic Analysis of CO2 Electrolysis Systems. Ind. Eng. Chem. Res. 2018, 57, 2165–2177. [Google Scholar] [CrossRef]
- Bushuyev, O.S.; De Luna, P.; Dinh, C.T.; Tao, L.; Saur, G.; van de Lagemaat, J.; Kelley, S.O.; Sargent, E.H. What Should We Make with CO2 and How Can We Make It? Joule 2018, 2, 825–832. [Google Scholar] [CrossRef] [Green Version]
- De Luna, P.; Hahn, C.; Higgins, D.; Jaffer, S.A.; Jaramillo, T.F.; Sargent, E.H. What Would It Take for Renewably Powered Electrosynthesis to Displace Petrochemical Processes? Science 2019, 364, eaav3506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, W.A.; Burdyny, T.; Vermaas, D.A.; Geerlings, H. Pathways to Industrial-Scale Fuel Out of Thin Air from CO2 Electrolysis. Joule 2019, 3, 1822–1834. [Google Scholar] [CrossRef]
- Keith, D.W.; Holmes, G.; Angelo, D.S.; Heidel, K. A Process for Capturing CO2 from the Atmosphere. Joule 2018, 2, 1573–1594. [Google Scholar] [CrossRef] [Green Version]
- Nitopi, S.; Bertheussen, E.; Scott, S.B.; Liu, X.; Engstfeld, A.K.; Horch, S.; Seger, B.; Stephens, I.E.L.; Chan, K.; Hahn, C.; et al. Progress and Perspectives of Electrochemical CO2 Reduction on Copper in Aqueous Electrolyte. Chem. Rev. 2019, 119, 7610–7672. [Google Scholar] [CrossRef] [Green Version]
- Torbensen, K.; Boudy, B.; Joulié, D.; von Wolff, N.; Robert, M. Emergence of CO2 Electrolyzers Including Supported Molecular Catalysts. Curr. Opin. Electrochem. 2020, 24, 49–55. [Google Scholar] [CrossRef]
- Liu, X.; Xiao, J.; Peng, H.; Hong, X.; Chan, K.; Nørskov, J.K. Understanding Trends in Electrochemical Carbon Dioxide Reduction Rates. Nat. Commun. 2017, 8, 15438. [Google Scholar] [CrossRef] [Green Version]
- Manbeck, G.F.; Fujita, E. A Review of Iron and Cobalt Porphyrins, Phthalocyanines and Related Complexes for Electrochemical and Photochemical Reduction of Carbon Dioxide. J. Porphyr. Phthalocya. 2015, 19, 45–64. [Google Scholar] [CrossRef]
- Corbin, N.; Zeng, J.; Williams, K.; Manthiram, K. Heterogeneous Molecular Catalysts for Electrocatalytic CO2 Reduction. Nano Res. 2019, 12, 2093–2125. [Google Scholar] [CrossRef]
- Zhang, R.; Warren, J.J. Recent Developments in Metalloporphyrin Electrocatalysts for Reduction of Small Molecules: Strategies for Managing Electron and Proton Transfer Reactions. ChemSusChem 2021, 14, 293–302. [Google Scholar] [CrossRef] [PubMed]
- Usman, M.; Humayun, M.; Garba, M.D.; Ullah, L.; Zeb, Z.; Helal, A.; Suliman, M.H.; Alfaifi, B.Y.; Iqbal, N.; Abdinejad, M.; et al. Electrochemical Reduction of CO2: A Review of Cobalt Based Catalysts for Carbon Dioxide Conversion to Fuels. Nanomaterials 2021, 11, 2029. [Google Scholar] [CrossRef]
- Sun, L.; Reddu, V.; Fisher, A.C.; Wang, X. Electrocatalytic Reduction of Carbon Dioxide: Opportunities with Heterogeneous Molecular Catalysts. Energy Environ. Sci. 2020, 13, 374–403. [Google Scholar] [CrossRef]
- Wu, Y.; Liang, Y.; Wang, H. Heterogeneous Molecular Catalysts of Metal Phthalocyanines for Electrochemical CO2 Reduction Reactions. Acc. Chem. Res. 2021, 54, 3149–3159. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.; Diercks, C.S.; Zhang, Y.-B.; Kornienko, N.; Nichols, E.M.; Zhao, Y.; Paris, A.R.; Kim, D.; Yang, P.; Yaghi, O.M.; et al. Covalent Organic Frameworks Comprising Cobalt Porphyrins for Catalytic CO2 Reduction in Water. Science 2015, 349, 1208–1213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diercks, C.S.; Lin, S.; Kornienko, N.; Kapustin, E.A.; Nichols, E.M.; Zhu, C.; Zhao, Y.; Chang, C.J.; Yaghi, O.M. Reticular Electronic Tuning of Porphyrin Active Sites in Covalent Organic Frameworks for Electrocatalytic Carbon Dioxide Reduction. J. Am. Chem. Soc. 2018, 140, 1116–1122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, D.-G.; Qiu, T.; Guo, W.; Liang, Z.; Tabassum, H.; Xia, D.; Zou, R. Covalent Organic Framework-Based Materials for Energy Applications. Energy Environ. Sci. 2021, 14, 688–728. [Google Scholar] [CrossRef]
- Abe, T.; Imaya, H.; Yoshida, T.; Tokita, S.; Schlettwein, D.; Wöhrle, D.; Kaneko, M. Electrochemical CO2 Reduction Catalysed by Cobalt Octacyanophthalocyanine and Its Mechanism. J. Porphyr. Phthalocya. 1997, 1, 315–321. [Google Scholar] [CrossRef]
- Lieber, C.M.; Lewis, N.S. Catalytic Reduction of Carbon Dioxide at Carbon Electrodes Modified with Cobalt Phthalocyanine. J. Am. Chem. Soc. 1984, 106, 5033–5034. [Google Scholar] [CrossRef]
- Kapusta, S.; Hackerman, N. Carbon Dioxide Reduction at a Metal Phthalocyanine Catalyzed Carbon Electrode. J. Electrochem. Soc. 1984, 131, 1511–1514. [Google Scholar] [CrossRef]
- Zhu, M.; Ye, R.; Jin, K.; Lazouski, N.; Manthiram, K. Elucidating the Reactivity and Mechanism of CO2 Electroreduction at Highly Dispersed Cobalt Phthalocyanine. ACS Energy Lett. 2018, 3, 1381–1386. [Google Scholar] [CrossRef]
- Zhang, X.; Wu, Z.; Zhang, X.; Li, L.; Li, Y.; Xu, H.; Li, X.; Yu, X.; Zhang, Z.; Liang, Y.; et al. Highly Selective and Active CO2 Reduction Electrocatalysts Based on Cobalt Phthalocyanine/Carbon Nanotube Hybrid Structures. Nat. Commun. 2017, 8, 14675. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boutin, E.; Wang, M.; Lin, J.C.; Mesnage, M.; Mendoza, D.; Lassalle-Kaiser, B.; Hahn, C.; Jaramillo, T.F.; Robert, M. Aqueous Electrochemical Reduction of Carbon Dioxide and Carbon Monoxide into Methanol with Cobalt Phthalocyanine. Angew. Chem. Int. Ed. 2019, 58, 16172–16176. [Google Scholar] [CrossRef]
- Wu, Y.; Jiang, Z.; Lu, X.; Liang, Y.; Wang, H. Domino Electroreduction of CO2 to Methanol on a Molecular Catalyst. Nature 2019, 575, 639–642. [Google Scholar] [CrossRef]
- Hu, X.-M.; Rønne, M.H.; Pedersen, S.U.; Skrydstrup, T.; Daasbjerg, K. Enhanced Catalytic Activity of Cobalt Porphyrin in CO2 Electroreduction upon Immobilization on Carbon Materials. Angew. Chem. Int. Ed. 2017, 56, 6468–6472. [Google Scholar] [CrossRef]
- Ren, S.; Joulié, D.; Salvatore, D.; Torbensen, K.; Wang, M.; Robert, M.; Berlinguette, C.P. Molecular Electrocatalysts Can Mediate Fast, Selective CO2 Reduction in a Flow Cell. Science 2019, 365, 367–369. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Torbensen, K.; Salvatore, D.; Ren, S.; Joulié, D.; Dumoulin, F.; Mendoza, D.; Lassalle-Kaiser, B.; Işci, U.; Berlinguette, C.P.; et al. CO2 Electrochemical Catalytic Reduction with a Highly Active Cobalt Phthalocyanine. Nat. Commun. 2019, 10, 3602. [Google Scholar] [CrossRef] [Green Version]
- Ghani, F.; Kristen, J.; Riegler, H. Solubility Properties of Unsubstituted Metal Phthalocyanines in Different Types of Solvents. J. Chem. Eng. Data 2012, 57, 439–449. [Google Scholar] [CrossRef]
- Snow, A.W. Phthalocyanine Aggregation. In The Porphyrin Handbook: Phthalocyanines: Properties and Materials; Elsevier Inc.: Amsterdam, The Netherlands, 2003; Volume 17, pp. 129–176. ISBN 9780123932273. [Google Scholar]
- Wu, Y.; Hu, G.; Rooney, C.L.; Brudvig, G.W.; Wang, H. Heterogeneous Nature of Electrocatalytic CO/CO2 Reduction by Cobalt Phthalocyanines. ChemSusChem 2020, 13, 6296–6299. [Google Scholar] [CrossRef]
- Durmuş, M.; Nyokong, T. Synthesis and Solvent Effects on the Electronic Absorption and Fluorescence Spectral Properties of Substituted Zinc Phthalocyanines. Polyhedron 2007, 26, 2767–2776. [Google Scholar] [CrossRef]
- Bayar, Ş.; Dınçer, H.A.; Uğur, A.L. Novel Non-Aggregated Soluble Metallophthalocyanines Bearing Carboxylic Acid Groups. J. Coord. Chem. 2012, 65, 1371–1380. [Google Scholar] [CrossRef]
- Biyiklioglu, Z.; Alp, H. Synthesis, Characterization, Electropolymerization and Aggregation Properties of Axially Diethyl-Dimethylaminophenoxypropanoxy Substituted Silicon Phthalocyanines and Their Water Soluble Derivatives. Dye. Pigment. 2016, 132, 213–222. [Google Scholar] [CrossRef]
- Choi, J.; Wagner, P.; Gambhir, S.; Jalili, R.; MacFarlane, D.R.; Wallace, G.G.; Officer, D.L. Steric Modification of a Cobalt Phthalocyanine/Graphene Catalyst to Give Enhanced and Stable Electrochemical CO2 Reduction to CO. ACS Energy Lett. 2019, 4, 666–672. [Google Scholar] [CrossRef]
- McKearney, D.; Zhou, W.; Zellman, C.; Williams, V.E.; Leznoff, D.B. Facile Tuning of Strong Near-IR Absorption Wavelengths in Manganese(III) Phthalocyanines via Axial Ligand Exchange. Chem. Commun. 2019, 55, 6696–6699. [Google Scholar] [CrossRef] [PubMed]
- McKeown, N.B.; Chambrier, I.; Cook, M.J. Synthesis and Characterisation of Some 1,4,8,11,15,18,22,25-Octa-Alkyl- and 1,4,8,11,15,18-Hexa-Alkyl-22,25-Bis(Carboxypropyl)Phthalocyanines. J. Chem. Soc., Perkin Trans. 1 1990, 0, 1169–1177. [Google Scholar] [CrossRef]
- Furuyama, T.; Uchiyama, S.; Iwamoto, T.; Maeda, H.; Segi, M. Changes of Phthalocyanine Visible Color Caused by Near-IR Solvatochromism. J. Porphyr. Phthalocyanines 2018, 22, 88–94. [Google Scholar] [CrossRef]
- Roshea, D. Manipulating the Electronic Properties of Metallophthalocyanines through Interactions with the Ligand’s Aromatic Core; Simon Fraser University: Burnaby, BC, Canada, 2022. [Google Scholar]
- Tolbin, A.Y.; Pushkarev, V.E.; Sedova, M.V.; Maklakov, S.S.; Tomilova, L.G. Aggregation of Slipped-Cofacial Phthalocyanine J-Type Dimers: Spectroscopic and AFM Study. Spectrochim. Acta A Mol. Biomol. Spectrosc. 2018, 205, 335–340. [Google Scholar] [CrossRef]
- Balaban, T.S. Tailoring Porphyrins and Chlorins for Self-Assembly in Biomimetic Artificial Antenna Systems. Acc. Chem. Res. 2005, 38, 612–623. [Google Scholar] [CrossRef]
- Charalambidis, G.; Georgilis, E.; Panda, M.K.; Anson, C.E.; Powell, A.K.; Doyle, S.; Moss, D.; Jochum, T.; Horton, P.N.; Coles, S.J.; et al. A Switchable Self-Assembling and Disassembling Chiral System Based on a Porphyrin-Substituted Phenylalanine–Phenylalanine Motif. Nat. Commun. 2016, 7, 12657. [Google Scholar] [CrossRef]
- Muto, T.; Temma, T.; Kimura, M.; Hanabusa, K.; Shirai, H. Elongation of the π-System of Phthalocyanines by Introduction of Thienyl Substituents at the Peripheral β Positions. Synthesis and Characterization. J. Org. Chem. 2001, 66, 6109–6115. [Google Scholar] [CrossRef]
- Feng, Q.; Sun, Y.; Gu, X.; Dong, Z. Advances of Cobalt Phthalocyanine in Electrocatalytic CO2 Reduction to CO: A Mini Review. Electrocatalysis 2022, 13, 675–690. [Google Scholar] [CrossRef]
- Abe, T.; Taguchi, F.; Yoshida, T.; Tokita, S.; Schnurpfeil, G.; Wöhrle, D.; Kaneko, M. Electrocatalytic CO2 Reduction by Cobalt Octabutoxyphthalocyanine Coated on Graphite Electrode. J. Mol. Catal. A-Chem. 1996, 112, 55–61. [Google Scholar] [CrossRef]
- Durand, R.R.; Anson, F.C. Catalysis of Dioxygen Reduction at Graphite Electrodes by an Adsorbed Cobalt(II) Porphyrin. J. Electroanal. Chem. 1982, 134, 273–289. [Google Scholar] [CrossRef]
- Song, E.; Shi, C.; Anson, F.C. Comparison of the Behavior of Several Cobalt Porphyrins as Electrocatalysts for the Reduction of O2 at Graphite Electrodes. Langmuir 1998, 14, 4315–4321. [Google Scholar] [CrossRef]
- Kaminsky, C.J.; Weng, S.; Wright, J.; Surendranath, Y. Adsorbed Cobalt Porphyrins Act like Metal Surfaces in Electrocatalysis. Nat. Catal. 2022, 5, 430–442. [Google Scholar] [CrossRef]
- Kramer, W.W.; McCrory, C.C.L. Polymer Coordination Promotes Selective CO2 Reduction by Cobalt Phthalocyanine. Chem. Sci. 2016, 7, 2506–2515. [Google Scholar] [CrossRef] [Green Version]
- Simpson, A.J.; Brown, S.A. Purge NMR: Effective and Easy Solvent Suppression. J. Magn. Reson. 2005, 175, 340–346. [Google Scholar] [CrossRef]
- Liu, Y.; McCrory, C.C.L. Modulating the Mechanism of Electrocatalytic CO2 Reduction by Cobalt Phthalocyanine through Polymer Coordination and Encapsulation. Nat. Commun. 2019, 10, 1683. [Google Scholar] [CrossRef] [Green Version]
- Dunwell, M.; Yan, Y.; Xu, B. Understanding the Influence of the Electrochemical Double-Layer on Heterogeneous Electrochemical Reactions. Curr. Opin. Chem. Eng. 2018, 20, 151–158. [Google Scholar] [CrossRef]
- Uddin, M.S.; Tanaya Das, H.; Maiyalagan, T.; Elumalai, P. Influence of Designed Electrode Surfaces on Double Layer Capacitance in Aqueous Electrolyte: Insights from Standard Models. Appl. Surf. Sci. 2018, 449, 445–453. [Google Scholar] [CrossRef]
- Jinwan, C.; Jundong, W.; Jinling, H.; Naisheng, C. The Ring-Substituted Phthalocyanines and Its Metal Complexes: Crystal Structure of 1,4,8,11,15,18,22,25-Octabutoxyphthalocyaninatocopper. Chin. Sci. Bull. 2002, 47, 3. [Google Scholar]
- Yoshida, T.; Kamato, K.; Tsukamoto, M.; Iida, T.; Schlettwein, D.; Wöhrle, D.; Kaneko, M. Selective Electrocatalysis for CO2 Reduction in the Aqueous Phase Using Cobalt Phthalocyanine/Poly-4-Vinylpyridine Modified Electrodes. J. Electroanal. Chem. 1995, 385, 209–225. [Google Scholar] [CrossRef]
- Costentin, C.; Drouet, S.; Robert, M.; Savéant, J.-M. Turnover Numbers, Turnover Frequencies, and Overpotential in Molecular Catalysis of Electrochemical Reactions. Cyclic Voltammetry and Preparative-Scale Electrolysis. J. Am. Chem. Soc. 2012, 134, 11235–11242. [Google Scholar] [CrossRef] [PubMed]
- Costentin, C.; Drouet, S.; Robert, M.; Savéant, J.-M. Correction to Turnover Numbers, Turnover Frequencies, and Overpotential in Molecular Catalysis of Electrochemical Reactions. Cyclic Voltammetry and Preparative-Scale Electrolysis. J. Am Chem. Soc. 2012, 134, 19949–19950. [Google Scholar] [CrossRef]
- Lee, K.J.; Elgrishi, N.; Kandemir, B.; Dempsey, J.L. Electrochemical and Spectroscopic Methods for Evaluating Molecular Electrocatalysts. Nat. Rev. Chem. 2017, 1, 1–4. [Google Scholar] [CrossRef]
- Wang, V.C.-C.; Johnson, B.A. Interpreting the Electrocatalytic Voltammetry of Homogeneous Catalysts by the Foot of the Wave Analysis and Its Wider Implications. ACS Catal. 2019, 9, 7109–7123. [Google Scholar] [CrossRef]
- Matheu, R.; Neudeck, S.; Meyer, F.; Sala, X.; Llobet, A. Foot of the Wave Analysis for Mechanistic Elucidation and Benchmarking Applications in Molecular Water Oxidation Catalysis. ChemSusChem 2016, 9, 3361–3369. [Google Scholar] [CrossRef] [PubMed]
- Costentin, C.; Savéant, J.-M. Multielectron, Multistep Molecular Catalysis of Electrochemical Reactions: Benchmarking of Homogeneous Catalysts. ChemElectroChem 2014, 1, 1226–1236. [Google Scholar] [CrossRef]
- Al-Raqa, S.Y. The Synthesis and Photophysical Properties of Novel, Symmetrical, Hexadecasubstituted Zn Phthalocyanines and Related Unsymmetrical Derivatives. Dye. Pigment. 2008, 77, 259–265. [Google Scholar] [CrossRef]
- Gao, D.; Xu, H.; Yan, T.; Peng, B. The Synthesis of Symmetrically Octa-Substituted Phthalocyanines and Their Physical and Photo-Physical Properties. J. Chin. Chem. Soc. 2001, 48, 1189–1196. [Google Scholar] [CrossRef]
- Attia, M.S.; Ali, K.; El-Kemary, M.; Darwish, W.M. Phthalocyanine-Doped Polystyrene Fluorescent Nanocomposite as a Highly Selective Biosensor for Quantitative Determination of Cancer Antigen 125. Talanta 2019, 201, 185–193. [Google Scholar] [CrossRef] [PubMed]
- Hanson, S.S.; Warren, J.J. Syntheses, Characterization, and Electrochemical Behavior of Alkylated 2-(2′-Quinolylbenzimidazole) Complexes of Rhenium (I). Can. J. Chem. 2017, 96, 119–123. [Google Scholar] [CrossRef]
- Rigsby, M.L.; Wasylenko, D.J.; Pegis, M.L.; Mayer, J.M. Medium Effects Are as Important as Catalyst Design for Selectivity in Electrocatalytic Oxygen Reduction by Iron–Porphyrin Complexes. J. Am. Chem. Soc. 2015, 137, 4296–4299. [Google Scholar] [CrossRef] [PubMed]
- Forsyth, T.P.; Bradley, D.; Williams, G.; Montalban, A.G.; Stern, C.L.; Barrett, A.G.M.; Hoffman, B.M. A Facile and Regioselective Synthesis of Trans-Heterofunctionalized Porphyrazine Derivatives. J. Org. Chem. 1998, 63, 331–336. [Google Scholar] [CrossRef]
- Cook, M.J.; Dunn, A.J.; Howe, S.D.; Thomson, A.J.; Harrison, K.J. Octa-Alkoxy Phthalocyanine and Naphthalocyanine Derivatives: Dyes with Q-Band Absorption in the Far Red or near Infrared. J. Chem. Soc. Perkin Trans. 1 1988, 8, 2453–2458. [Google Scholar] [CrossRef]
- Abdelkader, A.M.; Cooper, A.J.; Dryfe R a, W.; Kinloch, I.A. How to Get between the Sheets: A Review of Recent Works on the Electrochemical Exfoliation of Graphene Materials from Bulk Graphite. Nanoscale 2015, 7, 6944–6956. [Google Scholar] [CrossRef]
- Bard, A.J.; Faulkner LR Allen JBard Larry, R. Faulkner, Electrochemical Methods: Fundamentals and Applications, New York: Wiley, 2001, 2nd Ed. Russ. J. Electrochem. 2002, 38, 1364–1365. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | E1/2[ROPcCo+/0] a | E1/2[ROPcCo0/−] a |
|---|---|---|
| nBuOPcCo | −0.08 | −0.97 |
| secBuOPcCo | −0.14 | −0.94 |
| EtHexOPcCo | −0.14 | −0.94 |
| nBuOPcCo | ||||
|---|---|---|---|---|
| Deposition Solution Concentration | Loading (mol cm−2) | Average Γcat (mol cm−2 × 10−13) | kFOW (s−1) | Average CPE Current Density (μA cm−2) |
| 1000 µM | 5.5 × 10−8 | 3.2 ± 0.6 | 59 ± 20 | 76.7 |
| 180 µM | 9.9 × 10−9 | 4.3 ± 2.1 | 410 ± 290 | 83.0 |
| 32 µM | 1.8 × 10−9 | 4.3 ± 1.7 | 420 ± 70 | 153 |
| 5 µM | 2.8 × 10−10 | 7.1 ± 2.9 | 190 ± 60 | 73.6 |
| 1 µM | 5.6 × 10−11 | 3.8 ± 0.3 | 220 ± 90 | 90.3 |
| 0.18 µM | 1 × 10−11 | 0.23 ± 0.04 | 930 ± 150 | 8.7 |
| secBuOPcCo | ||||
| 1000 µM | 5.5 × 10−8 | 2.3 ± 0.6 | 4100 ± 1000 | 26.5 |
| 180 µM | 9.9 × 10−9 | 5.5 ± 0.1 | 810 ± 60 | 25.4 |
| 32 µM | 1.8 × 10−9 | 3.7 ± 0.7 | 550 ± 130 | 51.5 |
| 5 µM | 2.8 × 10−10 | 1.4 ± 0.2 | 2300 ± 1200 | 58.4 |
| 1 µM | 5.6 × 10−11 | -- a | -- a | 66.8 |
| 0.18 µM | 1 × 10−11 | -- a | -- a | 56.7 |
| EtHexOPcCo | ||||
| 1000 µM | 5.5 × 10−8 | 0.60 ± 0.12 | 2000 ± 570 | 135 |
| 180 µM | 9.9 × 10−9 | 3.6 ± 1.5 | 1800 ± 540 | 26.5 |
| 32 µM | 1.8 × 10−9 | 3.1 ± 0.5 | 730 ± 120 | 76.3 |
| 5 µM | 2.8 × 10−10 | 1.6 ± 0.3 | 1300 ± 160 | 66.2 |
| 1 µM | 5.6 × 10−11 | -- a | -- a | 48.6 |
| 0.18 µM | 1 × 10−11 | -- a | -- a | 57.9 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tajbakhsh, E.; McKearney, D.; Leznoff, D.B.; Warren, J.J. Heterogenous Preparations of Solution-Processable Cobalt Phthalocyanines for Carbon Dioxide Reduction Electrocatalysis. Inorganics 2023, 11, 43. https://doi.org/10.3390/inorganics11010043
Tajbakhsh E, McKearney D, Leznoff DB, Warren JJ. Heterogenous Preparations of Solution-Processable Cobalt Phthalocyanines for Carbon Dioxide Reduction Electrocatalysis. Inorganics. 2023; 11(1):43. https://doi.org/10.3390/inorganics11010043
Chicago/Turabian StyleTajbakhsh, Elahe, Declan McKearney, Daniel B. Leznoff, and Jeffrey J. Warren. 2023. "Heterogenous Preparations of Solution-Processable Cobalt Phthalocyanines for Carbon Dioxide Reduction Electrocatalysis" Inorganics 11, no. 1: 43. https://doi.org/10.3390/inorganics11010043