
Automated Screening and Filtering Scripts for GC×GC-TOFMS Metabolomics Data


Abstract
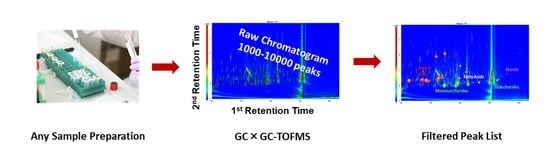
:
1. Introduction
2. Materials and Methods
2.1. Chemicals and Materials
2.1.1. Derivatization Materials
2.1.2. Standard Mixtures
2.2. Sample Preparation
2.2.1. Derivatization
2.2.2. SPME (Solid-Phase Microextraction)
2.3. GC×GC-TOFMS Analysis
2.4. Data Processing and Automated Classification
2.5. Scripting-Based Classifications and Evaluation
3. Results and Discussion
3.1. Evaluation of Scripts
3.2. Versatility of Scripts
3.3. Filtering of Peak Table by Scripts
3.4. Applying Cached Scripts
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bean, H.D.; Hill, J.E.; Dimandja, J.D. Improving the quality of biomarker candidates in untargeted metabolomics via peak table-based alignment of comprehensive two-dimensional gas chromatography–mass spectrometry data. J. Chromatogr. A 2015, 1394, 111–117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Higgins Keppler, E.A.; Jenkins, C.L.; Davis, T.J.; Bean, H.D. Advances in the application of comprehensive two-dimensional gas chromatography in metabolomics. TrAC Trends Anal. Chem. 2018, 109, 275–286. [Google Scholar] [CrossRef] [PubMed]
- Bilbao, A.; Gibbons, B.C.; Slysz, G.W.; Crowell, K.L.; Monroe, M.E.; Ibrahim, Y.M.; Smith, R.D.; Payne, S.H.; Baker, E.S. An algorithm to correct saturated mass spectrometry ion abundances for enhanced quantitation and mass accuracy in omic studies. Int. J. Mass Spectrom. 2018, 427, 91–99. [Google Scholar] [CrossRef] [Green Version]
- Almstetter, M.; Oefner, P.; Dettmer, K. Comprehensive two-dimensional gas chromatography in metabolomics. Anal. Bioanal.Chem. 2012, 402, 1993–2013. [Google Scholar] [CrossRef]
- Castillo, S.; Mattila, I.; Miettinen, J.; Orešič, M.; Hyötyläinen, T. Data Analysis Tool for Comprehensive Two-Dimensional Gas Chromatography/Time-of-Flight Mass Spectrometry. Anal. Chem. 2011, 83, 3058–3067. [Google Scholar] [CrossRef]
- Vaz-Freire, L.T.; da Silva, M.D.R.G.; Freitas, A.M.C. Comprehensive two-dimensional gas chromatography for fingerprint pattern recognition in olive oils produced by two different techniques in Portuguese olive varieties Galega Vulgar, Cobrançosa e Carrasquenha. Anal. Chim. Acta 2009, 633, 263–270. [Google Scholar] [CrossRef]
- Vasquez, N.P.; Crosnier de bellaistre-Bonose, M.; Lévêque, N.; Thioulouse, E.; Doummar, D.; Billette de Villemeur, T.; Rodriguez, D.; Couderc, R.; Robin, S.; Courderot-Masuyer, C.; et al. Advances in the metabolic profiling of acidic compounds in children’s urines achieved by comprehensive two-dimensional gas chromatography. J. Chromatogr. B 2015, 1002, 130–138. [Google Scholar] [CrossRef]
- de la Mata, A.P.; McQueen, R.H.; Nam, S.L.; Harynuk, J.J. Comprehensive two-dimensional gas chromatographic profiling and chemometric interpretation of the volatile profiles of sweat in knit fabrics. Anal. Bioanal. Chem. 2017, 409, 1905–1913. [Google Scholar] [CrossRef]
- Das, M.K.; Bishwal, S.C.; Das, A.; Dabral, D.; Varshney, A.; Badireddy, V.K.; Nanda, R. Investigation of Gender-Specific Exhaled Breath Volatome in Humans by GCxGC-TOF-MS. Anal. Chem. 2014, 86, 1229–1237. [Google Scholar] [CrossRef] [PubMed]
- Phillips, M.; Cataneo, R.N.; Chaturvedi, A.; Kaplan, P.D.; Libardoni, M.; Mundada, M.; Patel, U.; Zhang, X. Detection of an extended human volatome with comprehensive two-dimensional gas chromatography time-of-flight mass spectrometry. PLoS ONE 2013, 8, e75274. [Google Scholar] [CrossRef]
- Winnike, J.H.; Wei, X.; Knagge, K.J.; Colman, S.D.; Gregory, S.G.; Zhang, X. Comparison of GC-MS and GC×GC-MS in the Analysis of Human Serum Samples for Biomarker Discovery. J. Proteome Res. 2015, 14, 1810–1817. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murray, J.A. Qualitative and quantitative approaches in comprehensive two-dimensional gas chromatography. J. Chromatogr. A 2012, 1261, 58–68. [Google Scholar] [CrossRef] [PubMed]
- Seeley, J.V.; Seeley, S.K. Multidimensional Gas Chromatography: Fundamental Advances and New Applications. Anal. Chem. 2013, 85, 557–578. [Google Scholar] [CrossRef] [PubMed]
- Koek, M.M.; Jellema, R.H.; van der Greef, J.; Tas, A.C.; Hankemeier, T. Quantitative metabolomics based on gas chromatography mass spectrometry: Status and perspectives. Metabolomics 2010, 7, 307–328. [Google Scholar] [CrossRef] [Green Version]
- Kind, T.; Wohlgemuth, G.; Lee, D.Y.; Lu, Y.; Palazoglu, M.; Shahbaz, S.; Fiehn, O. FiehnLib: Mass Spectral and Retention Index Libraries for Metabolomics Based on Quadrupole and Time-of-Flight Gas Chromatography/Mass Spectrometry. Anal. Chem. 2009, 81, 10038–10048. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jennerwein, M.K.; Eschner, M.; Gröger, T.; Wilharm, T.; Zimmermann, R. Complete Group-Type Quantification of Petroleum Middle Distillates Based on Comprehensive Two-Dimensional Gas Chromatography Time-of-Flight Mass Spectrometry (GC×GC-TOFMS) and Visual Basic Scripting. Energy Fuels 2014, 28, 5670–5681. [Google Scholar] [CrossRef] [Green Version]
- Hilton, D.C.; Jones, R.S.; Sjödin, A. A method for rapid, non-targeted screening for environmental contaminants in household dust. J. Chromatogr. A 2010, 1217, 6851–6856. [Google Scholar] [CrossRef]
- Pena-Abaurrea, M.; Jobst, K.J.; Ruffolo, R.; Shen, L.; McCrindle, R.; Helm, P.A.; Reiner, E.J. Identification of Potential Novel Bioaccumulative and Persistent Chemicals in Sediments from Ontario (Canada) Using Scripting Approaches with GC×GC-TOF MS Analysis. Environ. Sci. Technol. 2014, 48, 9591–9599. [Google Scholar] [CrossRef]
- Hashimoto, S.; Takazawa, Y.; Fushimi, A.; Tanabe, K.; Shibata, Y.; Ieda, T.; Ochiai, N.; Kanda, H.; Ohura, T.; Tao, Q.; et al. Global and selective detection of organohalogens in environmental samples by comprehensive two-dimensional gas chromatography–tandem mass spectrometry and high-resolution time-of-flight mass spectrometry. J. Chromatogr. A 2011, 1218, 3799–3810. [Google Scholar] [CrossRef]
- S Haglund, P.; Löfstrand, K.; Siek, K.; Asplund, L. Powerful GC-TOF-MS Techniques for Screening, Identification and Quantification of Halogenated Natural Products. Mass Spectrom. 2013, 2, S0018. [Google Scholar] [CrossRef] [Green Version]
- Weggler, B.A.; Gröger, T.; Zimmermann, R. Advanced scripting for the automated profiling of two-dimensional gas chromatography-time-of-flight mass spectrometry data from combustion aerosol. J. Chromatogr. A 2014, 1364, 241–248. [Google Scholar] [CrossRef]
- Gómez, M.J.; Herrera, S.; Solé, D.; García-Calvo, E.; Fernández-Alba, A.R. Automatic Searching and Evaluation of Priority and Emerging Contaminants in Wastewater and River Water by Stir Bar Sorptive Extraction followed by Comprehensive Two-Dimensional Gas Chromatography-Time-of-Flight Mass Spectrometry. Anal. Chem. 2011, 83, 2638–2647. [Google Scholar] [CrossRef]
- Welthagen, W.; Schnelle-Kreis, J.; Zimmermann, R. Search criteria and rules for comprehensive two-dimensional gas chromatography–time-of-flight mass spectrometry analysis of airborne particulate matter. J. Chromatogr. A 2003, 1019, 233–249. [Google Scholar] [CrossRef]
- Reichenbach, S.E.; Kottapalli, V.; Ni, M.; Visvanathan, A. Computer language for identifying chemicals with comprehensive two-dimensional gas chromatography and mass spectrometry. J. Chromatogr. A 2005, 1071, 263–269. [Google Scholar] [CrossRef] [Green Version]
- Brokl, M.; Bishop, L.; Wright, C.G.; Liu, C.; McAdam, K.; Focant, J. Analysis of mainstream tobacco smoke particulate phase using comprehensive two-dimensional gas chromatography time-of-flight mass spectrometry. J. Sep. Sci. 2013, 36, 1037–1044. [Google Scholar] [CrossRef] [PubMed]
- Gröger, T.; Welthagen, W.; Mitschke, S.; Schäffer, M.; Zimmermann, R. Application of comprehensive two-dimensional gas chromatography mass spectrometry and different types of data analysis for the investigation of cigarette particulate matter. J. Sep. Sci. 2008, 31, 3366–3374. [Google Scholar] [CrossRef] [PubMed]
- Titaley, I.A.; Ogba, O.M.; Chibwe, L.; Hoh, E.; Cheong, P.H.-Y.; Simonich, S.L.M. Automating data analysis for two-dimensional gas chromatography/time-of-flight mass spectrometry non-targeted analysis of comparative samples. J. Chromatogr. A 2018, 1541, 57–62. [Google Scholar] [CrossRef] [PubMed]
- Halket, J.M.; Zaikin, V.G. Derivatization in Mass Spectrometry—1. Silylation. Eur. J. Mass Spectrom. 2003, 9, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Nicolescu, T.O. Interpretation of Mass Spectra. Mass Spectrom. 2017. [Google Scholar] [CrossRef] [Green Version]
- Mukhtar Abdul-Bari, M.; McQueen, R.H.; Paulina de la Mata, A.; Batcheller, J.C.; Harynuk, J.J. Retention and release of odorants in cotton and polyester fabrics following multiple soil/wash procedures. Text Res. J. 2020, 0040517520914411. [Google Scholar] [CrossRef]
- Nam, S.L.; de la Mata, A.P.; Dias, R.P.; Harynuk, J.J. Towards Standardization of Data Normalization Strategies to Improve Urinary Metabolomics Studies by GC×GC-TOFMS. Metabolites 2020, 10, 376. [Google Scholar] [CrossRef] [PubMed]
- Warwick B Dunn; David Broadhurst; Paul Begley; Eva Zelena; Sue Francis-mcintyre; Nadine Anderson; Marie Brown; Joshau D Knowles; Antony Halsall; John N Haselden; Andrew W Nicholls; Ian D Wilson; Douglas B Kell; Royston Goodacre Procedures for large-scale metabolic profiling of serum and plasma using gas chromatography and liquid chromatography coupled to mass spectrometry. Nat. Protoc. 2011, 6, 1060.
- Dixon, E.; Clubb, C.; Pittman, S.; Ammann, L.; Rasheed, Z.; Kazmi, N.; Keshavarzian, A.; Gillevet, P.; Rangwala, H.; Couch, R.D. Solid-phase microextraction and the human fecal VOC metabolome. PLoS ONE 2011, 6, e18471. [Google Scholar] [CrossRef] [PubMed] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Condition 1 | Condition 2 | |
|---|---|---|
| Primary Column | 30 m × 0.25 mm; 0.25 μm Rtx-5MS (Restek) | 30 m × 0.25 mm; 1.00 μm Rtx-5 (Restek) |
| Secondary Column | 1.7 m × 0.25 mm; 0.20 μm SLB-IL59 (Supelco) | 1.8 m × 0.18 mm; 0.18 μm Rtx-Wax (Restek) |
| GC×GC Method | Inlet temperature: 270 °C Carrier gas: helium, constant flow of 2 mL/min 70 °C (5 min), ramp at 9.7 °C/min to 280 °C (15 min) 2° oven: 10 °C offset to the GC oven Modulator: 10 °C off set to the 2° oven Transfer line: 270 °C Modulation: 2.0 s (0.4 hot, 0.6 cold) | Inlet temperature: 250 °C Carrier gas: helium, constant flow of 1.44 mL/min 80 °C (4 min), ramp at 3.5 °C/min to 230 °C (10 min) 2° oven: 5 °C offset to the GC oven Modulator: 15 °C offset to the 2° oven Transfer line: 250 °C Modulation: 2.5 s (0.6 hot, 0.65 cold) |
| MS Method | m/z 25–900 acquisition delay: 300 s acquisition rate: 200 spectra/second optimized voltage offset: 200 electron energy: −70 eV ion source: 200 °C | m/z 50–660 acquisition delay: 298 s acquisition rate: 200 spectra/second acquisition voltage: 1700 electron energy: −70 eV ion source: 200 °C |
| Total Analysis Time | 41.6 min | 56.9 min |
| Name | Classifications | 1tR, 2tR (s) | Quant S/N | Similarity | Reverse |
|---|---|---|---|---|---|
| d-Mannose, 2,3,4,5,6-pentakis-O-(trimethylsilyl)-, o-methyloxyme, (1Z)- | sugar_5TMS | 2375, 1.455 | 220 | 747 | 882 |
| d-Glucose, 2,3,4,5,6-pentakis-O-(trimethylsilyl)-, o-methyloxyme, (1Z)- | sugar_5TMS | 2382.5, 1.455 | 185 | 680 | 866 |
| d-Galactose, 2,3,4,5,6-pentakis-O-(trimethylsilyl)-, o-methyloxyme, (1E)- | sugar_5TMS | 2392.5, 1.445 | 381 | 823 | 887 |
| Palmitoleic acid 1TMS | monoenoicFA_TMS | 2775, 1.555 | 53 | 529 | 763 |
| trans-9-Octadecenoic acid, trimethylsilyl ester | monoenoicFA_TMS | 2912.5, 1.550 | 497 | 844 | 870 |
| 9,12-Octadecadienoic acid (Z, Z)-, trimethylsilyl ester | dienoicFA_TMS | 2905, 1.570 | 177 | 726 | 852 |
| Myristic acid, TMS derivative | SatFA_TMS | 2322.5, 1.560 | 348 | 789 | 855 |
| Dodecanoic acid, trimethylsilyl ester | SatFA_TMS | 1960, 1.585 | 863 | 860 | 912 |
| Trimethyl palmitate | SatFA_TMS | 2650, 1.545 | 3103 | 906 | 932 |
| Trimethyl stearate | SatFA_TMS | 2952.5, 1.535 | 1711 | 883 | 917 |
| Trimethylsilyl hexanoate | SatFA_TMS | 717.5, 1.605 | 2417 | 928 | 949 |
| Heptanoic acid TMS | SatFA_TMS | 922.5, 1.650 | 488 | 811 | 870 |
| Octanoic acid, trimethylsilyl ester | SatFA_TMS | 1137.5, 1.650 | 1037 | 914 | 932 |
| Trimethylsilyl nonanoate | SatFA_TMS | 1352.5, 1.640 | 3152 | 897 | 951 |
| Name | Classifications | 1tR, 2tR (s) | Quant S/N | Similarity | Reverse |
|---|---|---|---|---|---|
| L-Valine, N-(trimethylsilyl)-, trimethylsilyl ester | valine | 1022.5, 1.660 | 27,807 | 909 | 915 |
| L-Threonine, 3TMS derivative | threonine_3TMS | 1400, 1.635 | 13,900 | 937 | 938 |
| D-(+)-Turanose, octakis(trimethylsilyl) ether | sugar_8TMS | 3470, 1.525 | 13,774 | 848 | 852 |
| D-Lactose, octakis(trimethylsilyl) ether, methyloxime (isomer 2) | sugar_8TMS | 3555, 1.585 | 9779 | 912 | 912 |
| Maltose, octakis(trimethylsilyl) ether, methyloxime (isomer 2) | sugar_8TMS | 3627.5, 1.600 | 6295 | 913 | 917 |
| d-Glucose, 2,3,4,5,6-pentakis-O-(trimethylsilyl)-, o-methyloxyme, (1E)- | sugar_5TMS | 2372.5, 1.485 | 30,046 | 930 | 948 |
| d-Galactose, 2,3,4,5,6-pentakis-O-(trimethylsilyl)-, o-methyloxyme, (1E)- | sugar_5TMS | 2380, 1.485 | 16,393 | 924 | 941 |
| d-Glucose, 2,3,4,5,6-pentakis-O-(trimethylsilyl)-, o-methyloxyme, (1Z)- | sugar_5TMS | 2400, 1.540 | 19,742 | 936 | 955 |
| d-Galactose, 2,3,4,5,6-pentakis-O-(trimethylsilyl)-, o-methyloxyme, (1Z)- | sugar_5TMS | 2420, 1.530 | 10,441 | 949 | 969 |
| D-(-)-Fructose, pentakis(trimethylsilyl) ether, methyloxime (syn) | sugar_5TMS | 2367.5, 1.440 | 30,674 | 922 | 922 |
| D-Arabinose, tetrakis(trimethylsilyl) ether, ethyloxime (isomer 1) | sugar_4TMS | 1940, 1.480 | 24,484 | 916 | 916 |
| D-Arabinose, tetrakis(trimethylsilyl) ether, ethyloxime (isomer 2) | sugar_4TMS | 1957.5, 1.445 | 42,525 | 915 | 915 |
| D-Arabinose, tetrakis(trimethylsilyl) ether, ethyloxime (isomer 1) | sugar_4TMS | 1967.5, 1.480 | 30,778 | 919 | 919 |
| D-Arabinose, tetrakis(trimethylsilyl) ether, ethyloxime (isomer 1) | sugar_4TMS | 1995, 1.515 | 56 | 907 | 907 |
| Trimethylsilyl 2-[(Trimethylsilyl)amino]-3-[Trimethylsilyl)oxy] propanoate | serine_3TMS | 1332.5, 1.680 | 68 | 489 | 929 |
| Serine, 3TMS derivative | serine_3TMS | 1345, 1.660 | 21,854 | 933 | 934 |
| Phenylalanine, 2TMS derivative | phenylalanine_2TMS | 1900, 1.760 | 9406 | 923 | 924 |
| Linolenic acid, trimethylsilyl ester | multienoicFA_TMS | 2870, 1.605 | 4904 | 897 | 898 |
| à-Linolenic acid, TMS derivative | multienoicFA_TMS | 2907.5, 1.620 | 2745 | 885 | 886 |
| Arachidonic acid, TMS derivative | multienoicFA_TMS | 3122.5, 1.605 | 5790 | 918 | 918 |
| Eicosapentaenoic Acid, TMS derivative | multienoicFA_TMS | 3132.5, 1.610 | 6779 | 930 | 930 |
| Norlinolenicacid TMS | multienoicFA_TMS | 3155, 1.605 | 5041 | 839 | 856 |
| à-Linolenic acid, trimethylsilyl ester | multienoicFA_TMS | 3192.5, 1.605 | 5089 | 839 | 897 |
| Arachidonic acid, trimethylsilyl ester | multienoicFA_TMS | 3365, 1.610 | 4978 | 860 | 907 |
| Doconexent, TMS derivative | multienoicFA_TMS | 3375, 1.615 | 3783 | 914 | 914 |
| 7,10,13,16-Docosatetraenoic acid, (Z)-, TMS derivative | multienoicFA_TMS | 3390, 1.620 | 4778 | 896 | 897 |
| Eicosapentaenoic Acid, TMS derivative | multienoicFA_TMS | 3400, 1.625 | 4578 | 876 | 877 |
| Arachidonic acid, trimethylsilyl ester | multienoicFA_TMS | 3432.5, 1.595 | 54 | 581 | 769 |
| 9-Tetradecenoic acid, (E)-, TMS derivative | monoenoicFA_TMS | 2297.5, 1.595 | 9268 | 932 | 948 |
| 13-Methyltetradec-9-enoic acid, TMS derivative | monoenoicFA_TMS | 2465, 1.605 | 7970 | 813 | 855 |
| cis-9-Hexadecenoic acid, trimethylsilyl ester | monoenoicFA_TMS | 2612.5, 1.590 | 11,490 | 899 | 900 |
| cis-9-Hexadecenoic acid, trimethylsilyl ester | monoenoicFA_TMS | 2620, 1.575 | 12,246 | 910 | 910 |
| 10-Heptadecenoic acid, (Z)-, TMS derivative | monoenoicFA_TMS | 2767.5, 1.590 | 9970 | 876 | 877 |
| Trimethylsilyl (9E)-9-octadecenoate | monoenoicFA_TMS | 2907.5, 1.590 | 3780 | 920 | 937 |
| trans-9-Octadecenoic acid, trimethylsilyl ester | monoenoicFA_TMS | 2925, 1.570 | 10,067 | 912 | 929 |
| 11-Eicosenoic acid, (E)-, TMS derivative | monoenoicFA_TMS | 3190, 1.585 | 8552 | 883 | 898 |
| 13-Docosenoic acid, (Z)-, TMS derivative | monoenoicFA_TMS | 3452.5, 1.595 | 7640 | 891 | 892 |
| 15-Tetracosenoic acid, (Z)-, TMS derivative | monoenoicFA_TMS | 3695, 1.620 | 5686 | 874 | 876 |
| L-Leucine, N-(trimethylsilyl)-, trimethylsilyl ester | leucine_2tms | 1147.5, 1.660 | 18,073 | 903 | 905 |
| L-Isoleucine, N-(trimethylsilyl)-, trimethylsilyl ester | isoleucine_2TMS | 1197.5, 1.650 | 22,457 | 894 | 916 |
| Glycine, N, N-bis(trimethylsilyl)-, trimethylsilyl ester | glycine_TMS | 1225, 1.660 | 39,151 | 889 | 891 |
| Trimethylsilyl (9E,12E)-9,12-octadecadienoate | dienoicFA_TMS | 2897.5, 1.600 | 6814 | 923 | 947 |
| 9,12-Octadecadienoic acid (Z, Z)-, trimethylsilyl ester | dienoicFA_TMS | 2912.5, 1.555 | 4320 | 923 | 946 |
| 11,14-Eicosadienoic acid, TMS derivative | dienoicFA_TMS | 3182.5, 1.600 | 5420 | 890 | 908 |
| 13,16-Docasadienoic acid, (Z)-, TMS derivative | dienoicFA_TMS | 3447.5, 1.605 | 5594 | 853 | 854 |
| Silane, (dodecyloxy)trimethyl- | alcohol_TMS | 1792.5, 1.410 | 254 | 823 | 890 |
| Trimethylsilyl 2-[(trimethylsilyl)amino] propanoate | alanine_2TMS | 777.5, 1.625 | 19,604 | 910 | 916 |
| Bis (Trimethylsilyl) 2-[(Trimethylsilyl)amino] succinate | acidicAA | 1687.5, 1.835 | 16,566 | 882 | 914 |
| L-Aspartic acid, 3TMS derivative | acidicAA | 1690, 1.830 | 14,313 | 909 | 924 |
| L-Glutamic acid, 3TMS derivative | acidicAA | 1890, 1.855 | 4361 | 870 | 872 |
| Trimethyl stearate | SatFA_TMS | 2972.5, 1.535 | 138 | 648 | 854 |
| Docosanoic acid, trimethylsilyl ester | SatFA_TMS | 3517.5, 1.545 | 80 | 467 | 705 |
| Dodecanoic acid, trimethylsilyl ester | SatFA_TMS | 1957.5, 1.620 | 31,098 | 924 | 959 |
| Tridecanoic acid, TMS derivative | SatFA_TMS | 2140, 1.600 | 20,595 | 920 | 921 |
| Tetradecanoic acid, trimethylsilyl ester | SatFA_TMS | 2317.5, 1.590 | 23,511 | 921 | 939 |
| Pentadecanoic acid, TMS derivative | SatFA_TMS | 2485, 1.580 | 14,080 | 921 | 922 |
| Trimethyl palmitate | SatFA_TMS | 2645, 1.580 | 7568 | 928 | 963 |
| Heptadecanoic acid, TMS derivative | SatFA_TMS | 2800, 1.565 | 21,580 | 895 | 920 |
| Trimethyl stearate | SatFA_TMS | 2947.5, 1.570 | 9389 | 917 | 960 |
| Arachidic acid, TMS derivative | SatFA_TMS | 3225, 1.565 | 15,089 | 857 | 918 |
| Heneicosanoic acid, TMS derivative | SatFA_TMS | 3357.5, 1.570 | 15,982 | 841 | 843 |
| Docosanoic acid, trimethylsilyl ester | SatFA_TMS | 3482.5, 1.580 | 12,521 | 868 | 886 |
| Trimethylsilyl tricosanoate | SatFA_TMS | 3605, 1.590 | 13,168 | 846 | 861 |
| Trimethylsilyl ester of tetracosanoic acid | SatFA_TMS | 3722.5, 1.605 | 16,162 | 878 | 914 |
| Trimethylsilyl Hexanoate | SatFA_TMS | 717.5, 1.625 | 1416 | 917 | 933 |
| Heptanoic acid, TMS derivative | SatFA_TMS | 925, 1.660 | 446 | 842 | 878 |
| Octanoic acid, trimethylsilyl ester | SatFA_TMS | 1137.5, 1.670 | 1681 | 917 | 927 |
| Nonanoic acid, TMS derivative | SatFA_TMS | 1352.5, 1.660 | 4337 | 889 | 890 |
| 8-Methylnonanoic acid, trimethylsilyl ester | SatFA_TMS | 1562.5, 1.645 | 11,862 | 920 | 920 |
| Undecanoic acid, TMS derivative | SatFA_TMS | 1765, 1.625 | 166 | 631 | 778 |
| Methyl ç-linolenate | LinearTrienoicFAME | 2700, 1.665 | 2039 | 906 | 908 |
| 8,11,14-Eicosatrienoic acid, methyl ester, (Z, Z, Z)- | LinearTrienoicFAME | 3000, 1.660 | 2076 | 893 | 894 |
| Undecanoic acid, methyl ester | LinearSaturatedFAME | 1715, 1.705 | 236 | 653 | 837 |
| Tridecanoic acid, methyl ester | LinearSaturatedFAME | 1912.5, 1.685 | 1516 | 912 | 951 |
| Tetradecanoic acid, methyl ester | LinearSaturatedFAME | 2100, 1.670 | 11,892 | 935 | 938 |
| Pentadecanoic acid, methyl ester | LinearSaturatedFAME | 2280, 1.655 | 10,751 | 925 | 936 |
| Hexadecanoic acid, methyl ester | LinearSaturatedFAME | 2452.5, 1.640 | 35,352 | 930 | 930 |
| Heptadecanoic acid, methyl ester | LinearSaturatedFAME | 2620, 1.630 | 10,789 | 911 | 912 |
| Octadecanoic acid, methyl ester | LinearSaturatedFAME | 2777.5, 1.620 | 23,629 | 916 | 919 |
| Methyl icosanoate | LinearSaturatedFAME | 3072.5, 1.615 | 28,121 | 924 | 925 |
| Heneicosanoic acid, methyl ester | LinearSaturatedFAME | 3212.5, 1.615 | 14,766 | 909 | 912 |
| Docosanoic acid, methyl ester | LinearSaturatedFAME | 3345, 1.620 | 18,749 | 913 | 916 |
| Octadecanoic acid, methyl ester | LinearSaturatedFAME | 3475, 1.620 | 11,248 | 898 | 941 |
| Tetracosanoic acid, methyl ester | LinearSaturatedFAME | 3600, 1.630 | 19,398 | 921 | 940 |
| 5,8,11,14-Eicosatetraenoic acid, methyl ester, (all-Z)- | LinearMultienoicFAME | 2972.5, 1.655 | 1965 | 888 | 889 |
| Methyl myristoleate | LinearMonoenoicFAME | 2077.5, 1.695 | 1179 | 900 | 901 |
| 9-Octadecenoic acid (Z)-, methyl ester | LinearMonoenoicFAME | 2260, 1.675 | 2455 | 874 | 880 |
| cis-10-Heptadecenoic acid, methyl ester | LinearMonoenoicFAME | 2585, 1.655 | 4075 | 914 | 914 |
| 9-Octadecenoic acid (Z)-, methyl ester | LinearMonoenoicFAME | 2735, 1.645 | 8661 | 923 | 925 |
| cis-Methyl 11-eicosenoate | LinearMonoenoicFAME | 3035, 1.635 | 4759 | 893 | 893 |
| Cyclohexene, 1-butyl- | LinearDienoicFAME | 712.5, 1.410 | 198 | 653 | 777 |
| Naphthalene, decahydro-2-methyl- | LinearDienoicFAME | 827.5, 1.445 | 89 | 584 | 807 |
| 9,12-Octadecadienoic acid, methyl ester | LinearDienoicFAME | 2725, 1.660 | 4463 | 915 | 936 |
| 9,12-Octadecadienoic acid, methyl ester | LinearDienoicFAME | 2737.5, 1.615 | 2531 | 888 | 908 |
| cis-11,14-Eicosadienoic acid, methyl ester | LinearDienoicFAME | 3027.5, 1.645 | 4466 | 916 | 916 |
| FAMEs | Condition 1 | Condition 2 | Algae Extract |
|---|---|---|---|
| Total | 34 | 21 | 49 |
| Saturated | 15 | 11 | 18 |
| Monoenoic | 10 | 6 | 11 |
| Dienoic | 2 | 2 | 11 |
| Trienoic | 4 | 2 | 6 |
| Multienoic | 3 | 0 | 3 |
| Sample | SPME | Derivatization | ||
|---|---|---|---|---|
| Sweat | Feces | Plasma | Urine | |
| Total Number of Peaks | 3995 | 1685 | 5104 | 11,097 |
| Aldehydes | 12 (1) | 12 | 0 | 0 |
| 1° alcohols | 9 | 5 (2) | 0 | 0 |
| 2° alcohols | 5 | 3 | 0 | 0 |
| Ketones | 15 | 28 | 0 | 0 |
| Free fatty acids | 14 | 12 | 0 | 0 |
| FAMEs | 5 (1) | 0 (1) | 3 | 0 |
| FAEEs | 2 | 1 | 0 | 0 |
| Isopropylesters | 3 | 1 | 0 | 0 |
| Amino acids (TMS) | 0 | 0 | 8 | 18 |
| Free fatty acids (TMS) | 0 | 0 | 14 (1) | 4 |
| Sugars (TMS) | 0 | 0 | 3 | 25 |
| Sterol (TMS) | 0 | 0 | 2 | 1 |
| Others (TMS) | 0 | 0 | 0 | 5 |
| Total Classified Peaks | 65 (1) | 62 (3) | 30 (1) | 53 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nam, S.L.; de la Mata, A.P.; Harynuk, J.J. Automated Screening and Filtering Scripts for GC×GC-TOFMS Metabolomics Data. Separations 2021, 8, 84. https://doi.org/10.3390/separations8060084
Nam SL, de la Mata AP, Harynuk JJ. Automated Screening and Filtering Scripts for GC×GC-TOFMS Metabolomics Data. Separations. 2021; 8(6):84. https://doi.org/10.3390/separations8060084
Chicago/Turabian StyleNam, Seo Lin, A. Paulina de la Mata, and James J. Harynuk. 2021. "Automated Screening and Filtering Scripts for GC×GC-TOFMS Metabolomics Data" Separations 8, no. 6: 84. https://doi.org/10.3390/separations8060084