
A Validated UPLC-MS/MS Method for Rapid Quantification of Umifenovir in Plasma Samples and Its Greenness Assessment
 , , , and
, , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Stock Solution, Calibration Standard (CS) and Quality Control (QC) Sample Preparation
2.2. UPLC-MS/MS and Chromatographic Conditions
2.3. Sample Preparation
2.4. Method Validation
2.4.1. Selectivity and Sensitivity
2.4.2. Assay Linearity and Sensitivity
2.4.3. Accuracy and Precision
2.4.4. Extraction Recovery (ER) and Matrix Effects (ME)
2.4.5. Stability and Dilution Integrity
3. Results and Discussion
3.1. Method Development
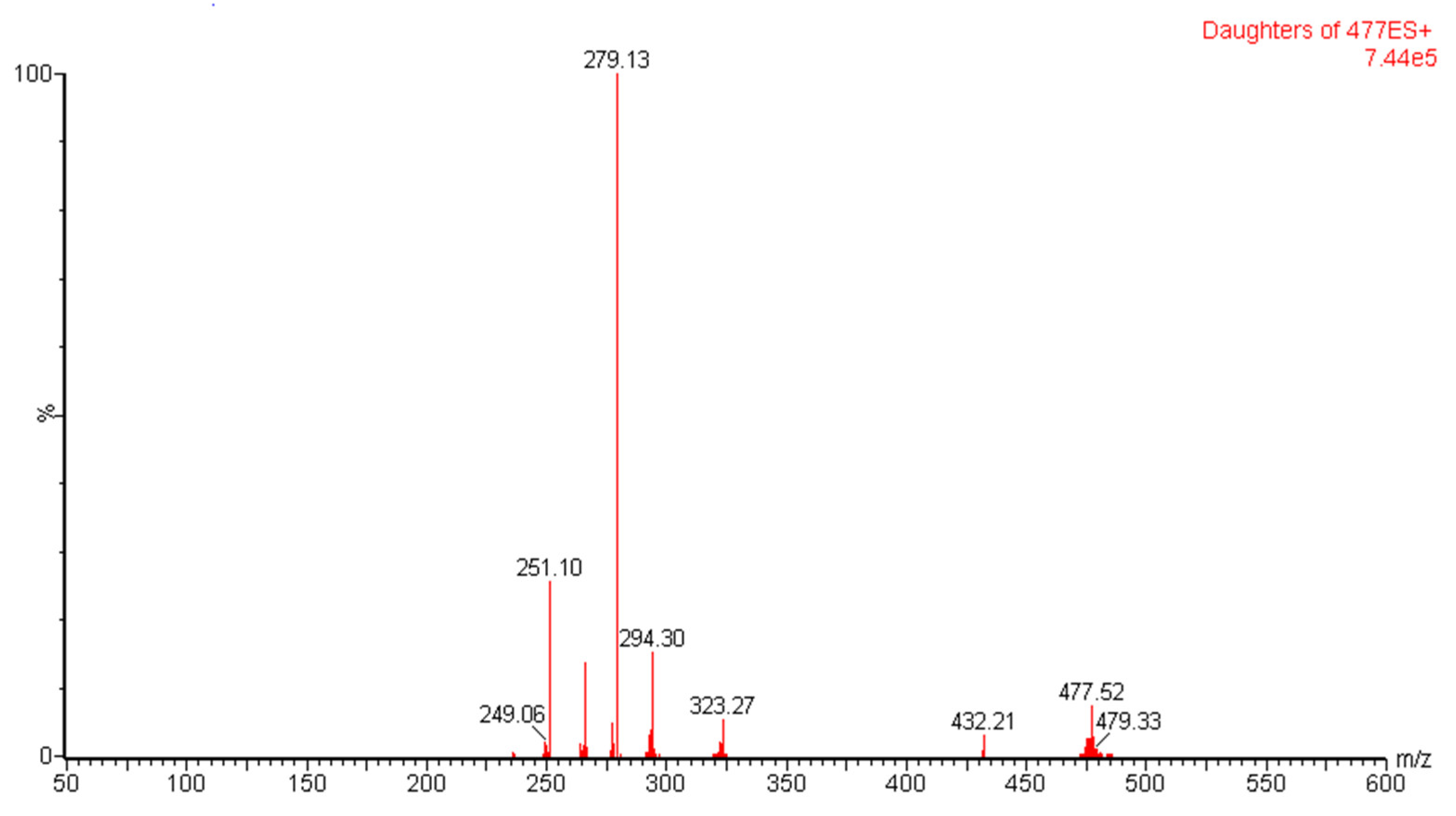
3.1.1. Optimization of Mass Spectroscopy Conditions
3.1.2. Optimization of Chromatographic Conditions
3.1.3. Optimization of Sample Preparation Method
3.2. Method Validation
3.2.1. Selectivity and Sensitivity
3.2.2. Linearity of the Method
3.2.3. Precision and Accuracy
3.2.4. Recovery and Matrix Effects
3.2.5. Stability and Dilution Integrity
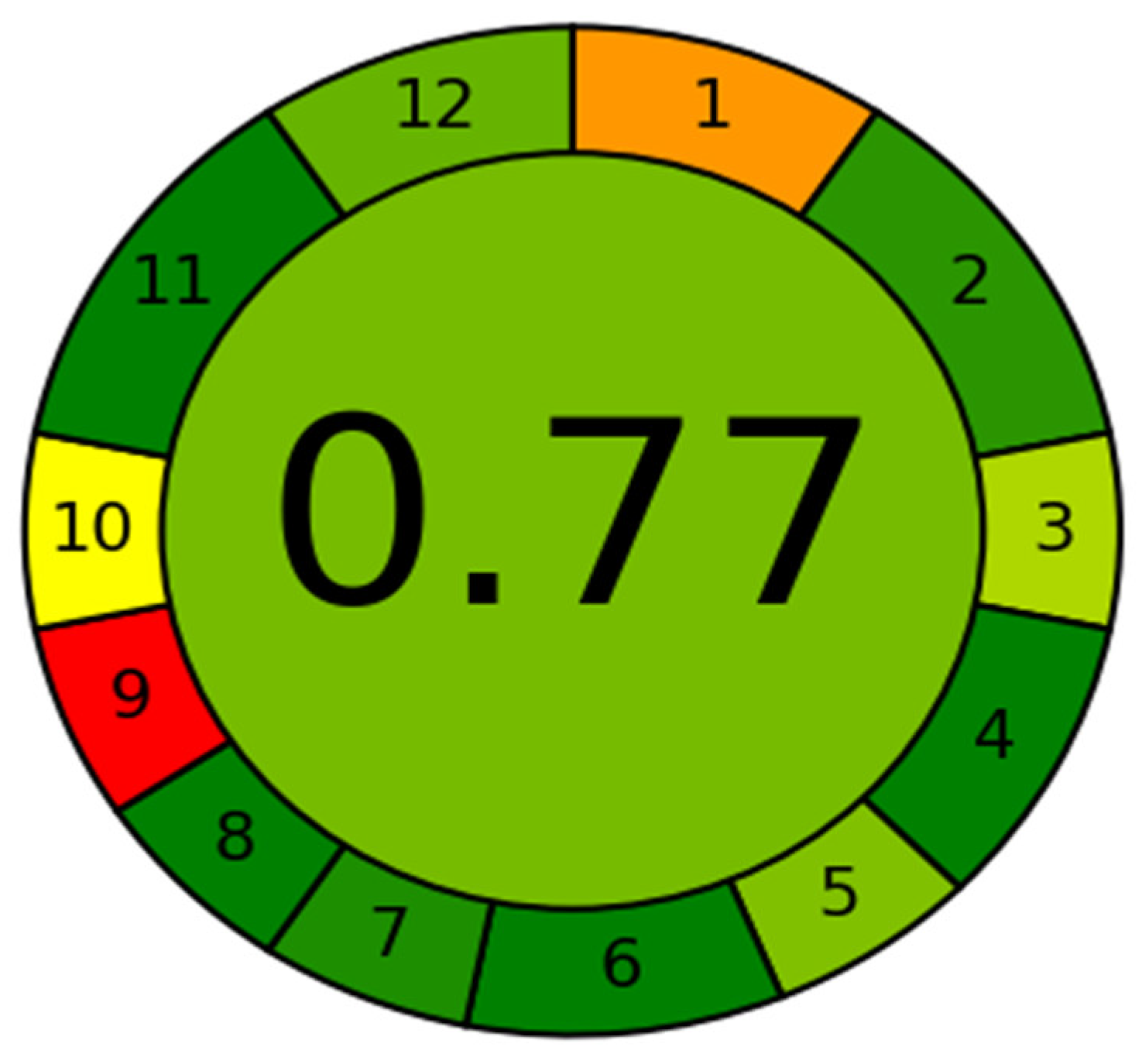
3.3. Greenness Assessment Using AGREE
3.4. Literature Comparison of the Method
3.5. Application in Pharmacokinetic Study of Novel Formulations in Rats
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Boriskin, Y.S.; Leneva, I.A.; Pécheur, E.I.; Polyak, S.J. Arbidol: A broad-spectrum antiviral compound that blocks viral fusion. Curr. Med. Chem. 2008, 15, 997–1005. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Xiong, H.R.; Lu, L.; Liu, Y.Y.; Luo, F.; Hou, W.; Yang, Z.Q. Antiviral and anti-inflammatory activity of arbidol hydrochloride in influenza A (H1N1) virus infection. Acta Pharmacol. Sin. 2013, 34, 1075–1083. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fink, S.L.; Vojtech, L.; Wagoner, J.; Slivinski, N.S.; Jackson, K.J.; Wang, R.; Khadka, S.; Luthra, P.; Basler, C.F.; Polyak, S.J. The antiviral drug arbidol inhibits Zika virus. Sci. Rep. 2018, 8, 8989. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pécheur, E.I.; Borisevich, V.; Halfmann, P.; Morrey, J.D.; Smee, D.F.; Prichard, M.; Mire, C.E.; Kawaoka, Y.; Geisbert, T.W.; Polyak, S.J. The synthetic antiviral drug arbidol inhibits globally prevalent pathogenic viruses. J. Virol. 2016, 90, 3086–3092. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pécheur, E.I.; Lavillette, D.; Alcaras, F.; Molle, J.; Boriskin, Y.S.; Roberts, M.; Cosset, F.L.; Polyak, S.J. Biochemical mechanism of hepatitis C virus inhibition by the broad- spectrum antiviral arbidol. Biochemistry 2007, 46, 6050–6059. [Google Scholar] [CrossRef] [Green Version]
- Blaising, J.; Polyak, S.J.; Pécheur, E.I. Arbidol as a broad spectrum antiviral: An update. Antivir. Res. 2014, 107, 84–94. [Google Scholar] [CrossRef]
- Zhonghua, L.; Xing, B.; Xue, Z. Special Expert Group for Control of the Epidemic of Novel Coronavirus Pneumonia of the Chinese Preventive Medicine Association. An update on the epidemiological characteristics of novel coronavirus pneumonia (COVID-19). Zhonghua Liu Xing Bing Xue Za Zhi 2020, 41, 139–144. [Google Scholar]
- Jie, X.; Hongmei, Y.; Ping, F.; Kuikui, Z.; Bohan, Y.; Rui, M. Beneficial effect of Arbidol in the management of COVID-19 infection. Aging 2021, 13, 9253–9264. [Google Scholar] [CrossRef]
- Gao, W.; Chen, S.; Wang, K.; Chen, R.; Guo, Q.; Lu, J.; Wu, X.; He, Y.; Yan, Q.; Wang, S.; et al. Clinical features and efficacy of antiviral drug, Arbidol In 220 nonemergency COVID-19 patients from East-West-Lake Shelter Hospital in Wuhan: A retrospective case series. Virol. J. 2020, 17, 162. [Google Scholar] [CrossRef]
- Nojomi, M.; Yassin, Z.; Keyvani, H.; Makiani, M.J.; Roham, M.; Laal, A.; Dehghan, N.; Navaei, M.; Ranjbar, M. Effect of Arbidol (Umifenovir) on COVID-19: A randomized controlled trial. BMC Infect. Dis. 2020, 20, 954. [Google Scholar] [CrossRef]
- Lin, L.; Li, T.S. Diagnosis and treatment plan for COVID-19 (trial version 8 revision) National Health Commission of the People’s Republic of China. Chin. J. Clin. Infect. Dis. 2021, 14, 81–88. [Google Scholar]
- Abdelrahman, Z.; Liu, Q.; Jiang, S.; Li, M.; Sun, Q.; Zhang, Y.; Wang, X. Evaluation of the current therapeutic approaches for COVID-19: A systematic review and a meta-analysis. Front. Pharmacol. 2021, 12, 607408. [Google Scholar] [CrossRef]
- Jomah, S.; Asdaq, S.M.B.; Al-Yamani, M.J. Clinical efficacy of antivirals against novel coronavirus (COVID-19): A review. J. Infect. Public Health 2020, 13, 1187–1195. [Google Scholar] [CrossRef]
- Ul’yanovskii, N.V.; Kosyakov, D.S.; Sypalov, S.A.; Varsegov, I.S.; Shavrina, I.S.; Lebedev, A.T. Antiviral drug Umifenovir (Arbidol) in municipal wastewater during the COVID-19 pandemic: Estimated levels and transformation. Sci. Total Environ. 2022, 805, 150380. [Google Scholar] [CrossRef]
- Full Prescribing Information: Arbidol (umifenovir) film-coated tablets 50 and 100 mg: Corrections and Additions. State Register of Medicines. Open joint-stock company “Pharmstandard-Tomskchempharm Prospekt Lenina, Tomsk, Tomsk Region, 634009”. Available online: https://newdrugapprovals.org/2020/03/21/arbidol-umifenovir/ (accessed on 15 April 2023). (In Russian).
- Liu, X.; Chen, X.H.; Zhang, Y.Y.; Liu, W.T.; Bi, K.S. Determination of arbidol in rat plasma by HPLC-UV using cloud-point extraction. J. Chromatogr. B 2007, 856, 273–277. [Google Scholar] [CrossRef]
- Metz, R.; Muth, P.; Ferger, M.; Kukes, V.G.; Vergin, H. Sensitive high-performance liquid chromatographic determination of arbidol, a new antiviral compound, in human plasma. J. Chromatogr. A 1998, 810, 63–69. [Google Scholar] [CrossRef]
- Xiong, X.; Zhai, S. High-performance liquid chromatography/tandem mass spectrometry method for the determination of arbidol in human plasma. J. AOAC Int. 2011, 94, 1100–1105. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Huang, Y.W.; Li, J.; Li, X.; Bi, K.S.; Chen, X.H. Determination of arbidol in human plasma by LC-ESI-MS. J. Pharm. Biomed. Anal. 2007, 43, 371–375. [Google Scholar] [CrossRef]
- Iqbal, M.; Ezzeldin, E.; Anwer, M.K.; Imam, F. Eco-Friendly UPLC-MS/MS Quantitation of Delafloxacin in Plasma and Its Application in a Pharmacokinetic Study in Rats. Separations 2021, 8, 146. [Google Scholar] [CrossRef]
- Alam, P.; Ezzeldin, E.; Iqbal, M.; Mostafa, G.A.E.; Anwer, M.K.; Alqarni, M.H.; Foudah, A.I.; Shakeel, F. Determination of Delafloxacin in Pharmaceutical Formulations Using a Green RP-HPTLC and NP-HPTLC Methods: A Comparative Study. Antibiotics 2020, 9, 359. [Google Scholar] [CrossRef]
- Alam, P.; Ezzeldin, E.; Iqbal, M.; Anwer, M.K.; Mostafa, G.A.; Alqarni, M.H.; Foudah, A.I.; Shakeel, F. Ecofriendly densitometric RP-HPTLC method for determination of rivaroxaban in nanoparticle formulations using green solvents. RSC Adv. 2020, 10, 2133–2140. [Google Scholar] [CrossRef] [Green Version]
- Haq, N.; Iqbal, M.; Alanazi, F.K.; Alsarra, I.A.; Shakeel, F. Applying green analytical chemistry for rapid analysis of drugs Adding health to pharmaceutical industry. Arab. J. Chem. 2017, 10, S777–S785. [Google Scholar] [CrossRef] [Green Version]
- Iqbal, M. UHPLC-MS/MS assay using environment friendly organic solvents: A green approach for fast determination of quetiapine in rat plasma. Arab. J. Chem. 2019, 12, 1774–1782. [Google Scholar] [CrossRef] [Green Version]
- Ibrahim, F.A.; Elmansi, H.; Fathy, M.E. Green RP-HPLC method for simultaneous determination of moxifloxacin combinations: Investigation of the greenness for the proposed method. Microchem. J. 2019, 148, 151–161. [Google Scholar] [CrossRef]
- Abou-Taleb, N.H.; Al-Enany, N.M.; El-Sherbiny, D.T.; El-Subbagh, H.I. Digitally enhanced thin layer chromatography for simultaneous determination of norfloxacin tinidazole with the aid of Taguchi orthogonal array and desirability function approach: Greenness assessment by analytical eco-scale. J. Sep. Sci. 2020, 43, 1195–1202. [Google Scholar] [CrossRef] [PubMed]
- Pena-Pereira, F.; Wojnowski, W.; Tobiszewski, M. AGREE Analytical Greenness metric approach and software. Anal. Chem. 2020, 92, 10076–10082. [Google Scholar] [CrossRef]
- Duan, X.; Liu, X.; Dong, Y.; Yang, J.; Zhang, J.; He, S.; Yang, F.; Wang, Z.; Dong, Y. A green HPLC method for determination of nine sulfonamides in milk and beef, and its greenness assessment with analytical eco-scale and greenness profile. J AOAC Int. 2020, 103, 1181–1189. [Google Scholar] [CrossRef]
- Abdelrahman, M.M.; Abdelwahab, N.S.; Hegazy, M.A.; Fares, M.Y.; EL-Sayed, G.M. Determination of the abused intravenously administered madness drops (tropicamide) by liquid chromatography in rat plasma; an application to pharmacokinetic study and greenness profile assessment. Microchem. J. 2020, 159, E105582. [Google Scholar] [CrossRef]
- Bioanalytical Method Validation Guidence for Industry. Food and Drug Administration, Center for Drug Evaluation and Research. Center for Veterinary Medicine (CVM). 2018. Available online: https://www.fda.gov/files/drugs/published/Bioanalytical-Method-Validation-Guidance-for-Industry.pdf (accessed on 15 February 2023).
- Anwer, M.K.; Iqbal, M.; Ahmed, M.M.; Aldawsari, M.F.; Ansari, M.N.; Ezzeldin, E.; Khalil, N.Y.; Ali, R. Improving the Solubilization and Bioavailability of Arbidol Hydrochloride by the Preparation of Binary and Ternary β-Cyclodextrin Complexes with Poloxamer 188. Pharmaceuticals 2021, 14, 411. [Google Scholar] [CrossRef]
- Liu, M.Y.; Wang, S.; Yao, W.F.; Wu, H.Z.; Meng, S.N.; Wei, M.J. Pharmacokinetic properties and bioequivalence of two formulations of arbidol: An open-label, single-dose, randomized-sequence, two-period crossover study in healthy Chinese male volunteers. Clin. Ther. 2009, 31, 784–792. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Concentration (ng mL−1) | Intra-Day (n = 5) | Inter-Day (n = 15) | ||||
|---|---|---|---|---|---|---|
| Mean ± SD (ng mL−1) | Precision RSD (%) | Accuracy (%) | Mean ± SD (ng mL−1) | Precision RSD (%) | Accuracy (%) | |
| 1.32 | 1.39 ± 0.06 | 4.62 | 105.8 | 1.44 ± 0.16 | 11.21 | 108.9 |
| 4.0 | 3.91 ± 0.23 | 5.88 | 97.8 | 3.74 ± 0.32 | 8.49 | 93.5 |
| 110 | 113 ± 10.10 | 9.65 | 102.7 | 116 ± 8.32 | 7.14 | 105.9 |
| 555 | 498 ± 7.92 | 1.59 | 90.5 | 483 ± 25.5 | 5.29 | 87.8 |
| conc. (ng mL−1) | % ER | % ME | |||
|---|---|---|---|---|---|
| % Mean ± SD | % RSD | %Mean ± SD | % RSD | ||
| Umifenovir | 4.0 | 83.97 ± 8.50 | 13.70 | 93.56 ± 3.99 | 4.27 |
| 110 | 81.54 ± 5.19 | 6.37 | 97.04 ± 3.20 | 3.29 | |
| 550 | 76.35 ± 6.43 | 8.42 | 91.73 ± 3.48 | 3.79 | |
| average | 80.60 ± 3.89 | 4.83 | 94.11 ± 2.70 | 2.87 | |
| IS | 25 | 84.56 ± 6.26 | 7.43 | 89.08 ± 5.43 | 6.09 |
| Stability | Nominal Concentration (ng mL−1) (n = 6) | Precision (RSD, %) | Accuracy |
|---|---|---|---|
| (%) | |||
| Short-term (8 h) | |||
| 4 | 7.86 | 104.6 | |
| 550 | 3.25 | 97.7 | |
| Freeze–thaw (three cycles) | |||
| 4 | 2.93 | 108.9 | |
| 550 | 5.20 | 93.2 | |
| Auto-sampler (24 h) | |||
| 4 | 5.53 | 109.4 | |
| 550 | 7.19 | 97.8 | |
| 3 months at −80 °C | |||
| 4 | 8.05 | 91.6 | |
| 550 | 9.34 | 86.3 | |
| Dilution integrity | 2 times | 6.76 | 107.6 |
| 4 times | 8.64 | 102.4 |
| Method | Extraction Solvent (Volume) | Calibration Range (ng mL−1) | Run Time | Application | Reference |
|---|---|---|---|---|---|
| (min) | |||||
| HPLC-MS/MS | TBME | 0.5–500 | 6 | H1N1 influenza virus-infected patients | [18] |
| (0.6 mL) | |||||
| HPLC-U/V | surfactant Triton X-114 (1 mL) | 80–20,000 | 10 | Pharmacokinetic study in rats | [16] |
| LC-ESI-MS/MS | Diethyl ether (3 mL) | 1–1000 | 6 | Healthy men volunteers | [19] |
| HPLC-UV | TBME | 5–10,000 | 25 | Healthy men volunteers | [17] |
| (5 mL) | |||||
| This method | Ethyl acetate | 1.32–625 | 2.5 | Pharmacokinetic study in rats | - |
| (1 mL) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Iqbal, M.; Imam, F.; Ali, E.A.; Kalam, M.A.; Alhudaithi, S.S.; Anwer, M.K. A Validated UPLC-MS/MS Method for Rapid Quantification of Umifenovir in Plasma Samples and Its Greenness Assessment. Separations 2023, 10, 379. https://doi.org/10.3390/separations10070379
Iqbal M, Imam F, Ali EA, Kalam MA, Alhudaithi SS, Anwer MK. A Validated UPLC-MS/MS Method for Rapid Quantification of Umifenovir in Plasma Samples and Its Greenness Assessment. Separations. 2023; 10(7):379. https://doi.org/10.3390/separations10070379
Chicago/Turabian StyleIqbal, Muzaffar, Faisal Imam, Essam A. Ali, Mohd Abul Kalam, Sulaiman S. Alhudaithi, and Md. Khalid Anwer. 2023. "A Validated UPLC-MS/MS Method for Rapid Quantification of Umifenovir in Plasma Samples and Its Greenness Assessment" Separations 10, no. 7: 379. https://doi.org/10.3390/separations10070379