
Extraction of Organochlorine Pesticides from Porous Membrane Packed Dried Fish Samples: Method Development and Greenness Evaluation
Abstract
:1. Introduction
- The concentration of target compounds is too low to perform direct determination without enrichment.
- The matrix is complex, and needs clean-up before injecting into the instrument to avoid interference and instrument incompatibility issues.
- The chemical features of the target compounds mismatch with the available instrumentation, and thus a chemical conversion (derivatization) is required.
2. Experimental
2.1. Materials and Chemicals

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Chemical/Material | Manufacturer |
|---|---|
| OCPs standard (15 OCPs investigated in this study are listed in Table 2). | Restek (Bellefonte, PA, USA) |
| Polypropylene (PP) membrane sheet (pore size: 0.2; thickness of 157 μm) | Membrana (Wuppertal, Germany) |
| Solvents (chloroform, methanol, n-hexane, dichloromethane (DCM)) | Fisher (Loughborough, UK) |
| Fish samples | Local market (Al-khobar, KSA) |
| Analyte | Retention Time (min) | Selected m/z Values | ||
|---|---|---|---|---|
| Heptachlor | 11.9 | 100 | 272 | 274 |
| Aldrin | 13.0 | 66 | 263 | 79 |
| Heptachlor Epoxide | 14.6 | 81 | 353 | 355 |
| Trans-chlordane | 15.6 | 33 | 375 | 377 |
| Endosulfan I | 16.2 | 241 | 239 | 195 |
| Cis-chlordane | 16.3 | 373 | 375 | 377 |
| Dieldrin | 17.5 | 79 | 81 | 82 |
| 4,4′-DDE | 17.7 | 246 | 318 | 248 |
| Endrin | 18.6 | 81 | 79 | 263 |
| Endosulfan II | 19.2 | 195 | 241 | 237 |
| 4,4′-DDD | 20.1 | 235 | 237 | 165 |
| Endrin Aldehyde | 20.2 | 67 | 345 | 250 |
| Endosulfan Sulfate | 21.5 | 387 | 272 | 274 |
| 4,4′-DDT | 21.9 | 235 | 23 | 165 |
| Methoxychlor | 24.8 | 227 | 228 | - |
2.2. Preparation of Fish Samples
2.3. Extraction Procedure
2.4. Optimization of Extraction Parameters
2.5. GC-MS Analysis
2.6. Analytical Method Validation
2.7. Greenness Evaluation
3. Results and Discussion
3.1. Optimization of Extraction Parameters
3.1.1. Extraction Solvent
3.1.2. Solvent Volume
3.1.3. Amount of Sample
3.1.4. Ultrasound Intensity
3.1.5. Extraction Time
3.2. Analytical Method Validation
3.3. Relative Recoveries, Analysis of Real Samples and Comparison with Other Methods
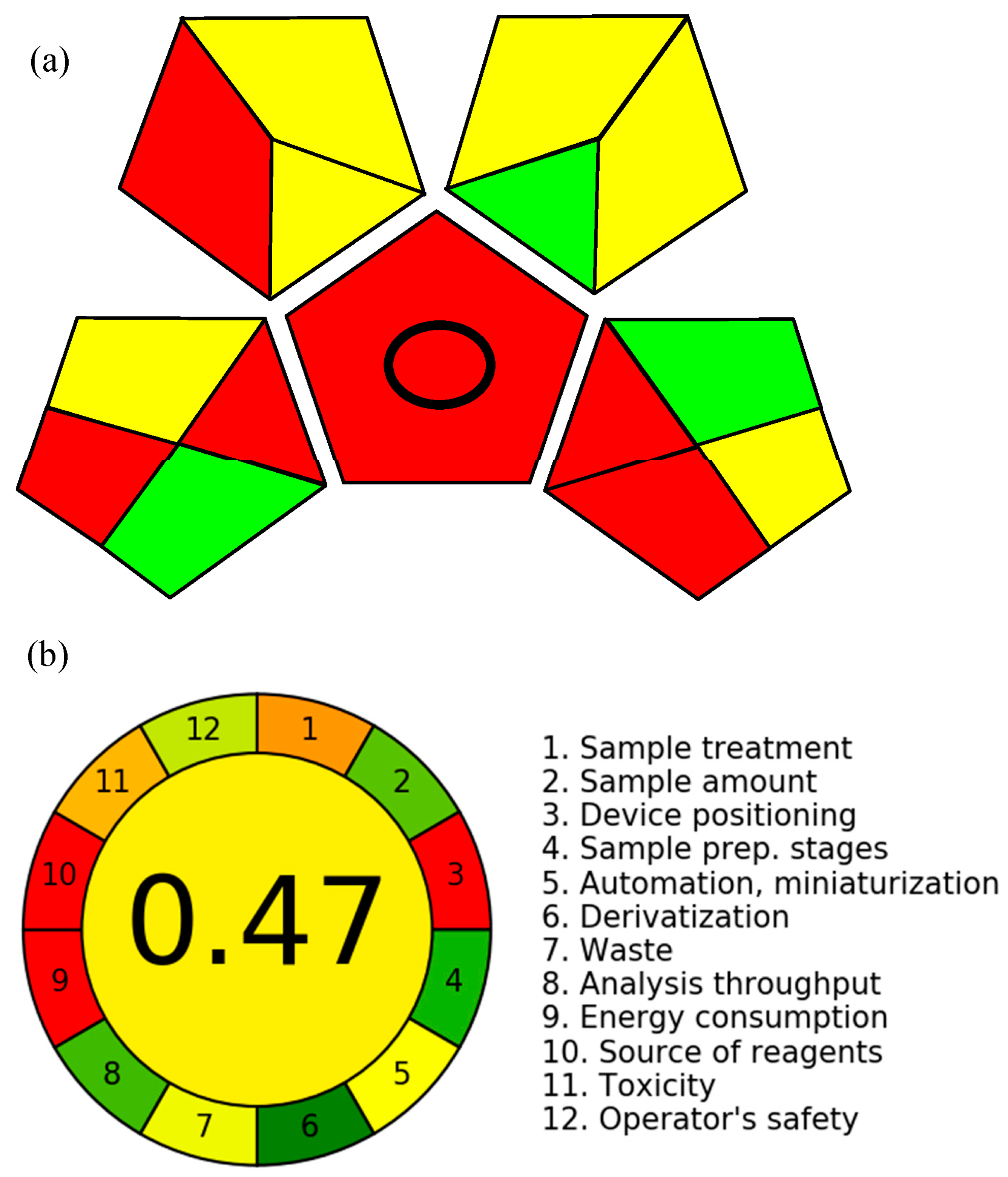
4. Greenness Evaluation
- (i)
- Sample collection;
- (ii)
- Sample transportation;
- (iii)
- Type of method;
- (iv)
- Solvents employed;
- (v)
- Energy related to instrumentation;
- (vi)
- Waste treatment.
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chisvert, A.; Cárdenas, S.; Lucena, R. Dispersive Micro-Solid Phase Extraction. TrAC-Trends Anal. Chem. 2019, 112, 226–233. [Google Scholar] [CrossRef]
- Sajid, M.; Kalinowska, K.; Płotka-Wasylka, J. Ferrofluids Based Analytical Extractions and Evaluation of Their Greenness. J. Mol. Liq. 2021, 339, 116901. [Google Scholar] [CrossRef]
- Sajid, M.; Nazal, M.K.; Ihsanullah, I. Novel Materials for Dispersive (Micro) Solid-Phase Extraction of Polycyclic Aromatic Hydrocarbons in Environmental Water Samples: A Review. Anal. Chim. Acta 2021, 1141, 246–262. [Google Scholar] [CrossRef] [PubMed]
- Gutiérrez-Serpa, A.; González-Martín, R.; Sajid, M.; Pino, V. Greenness of Magnetic Nanomaterials in Miniaturized Extraction Techniques: A Review. Talanta 2021, 225, 122053. [Google Scholar] [CrossRef] [PubMed]
- Płotka-Wasylka, J.; Owczarek, K.; Namieśnik, J. Modern Solutions in the Field of Microextraction Using Liquid as a Medium of Extraction. TrAC-Trends Anal. Chem. 2016, 85, 46–64. [Google Scholar] [CrossRef]
- Płotka-Wasylka, J.; Szczepańska, N.; de la Guardia, M.; Namieśnik, J. Miniaturized Solid-Phase Extraction Techniques. TrAC-Trends Anal. Chem. 2015, 73, 19–38. [Google Scholar] [CrossRef]
- Sajid, M. Dispersive Liquid-Liquid Microextraction: Evolution in Design, Application Areas, and Green Aspects. TrAC-Trends Anal. Chem. 2022, 152, 116636. [Google Scholar] [CrossRef]
- Yamini, Y.; Rezazadeh, M.; Seidi, S. Liquid-Phase Microextraction—The Different Principles and Configurations. TrAC-Trends Anal. Chem. 2019, 112, 264–272. [Google Scholar] [CrossRef]
- Picó, Y. Recent Advances in Sample Preparation for Pesticide Analysis. Compr. Sampl. Sample Prep. Anal. Tech. Sci. 2012, 3, 569–590. [Google Scholar] [CrossRef]
- Kataoka, H. Column-Switching Sample Preparation. Compr. Sampl. Sample Prep. Anal. Tech. Sci. 2012, 2, 649–676. [Google Scholar] [CrossRef]
- Soares, J.B.; Medina, D.A.V.; Toffoli, A.L.; Maciel, E.V.M.; Lanças, F.M. Recent Advances and Trends in Miniaturized Sample Preparation Techniques. J. Sep. Sci. 2020, 43, 202–225. [Google Scholar] [CrossRef] [PubMed]
- Arthur, C.L.; Pawliszyn, J. Solid Phase Microextraction with Thermal Desorption Using Fused Silica Optical Fibers. Anal. Chem. 1990, 62, 2145–2148. [Google Scholar] [CrossRef]
- Liu, S.; Dasgupta, P.K. Liquid Droplet. A Renewable Gas Sampling Interface. Anal. Chem. 2002, 67, 2042–2049. [Google Scholar] [CrossRef]
- Liu, H.; Dasgupta, P.K. Analytical Chemistry in a Drop. Solvent Extraction in a Microdrop. Anal. Chem. 1996, 68, 1817–1821. [Google Scholar] [CrossRef] [PubMed]
- Jeannot, M.A.; Cantwell, F.F. Solvent Microextraction into a Single Drop. Anal. Chem. 1996, 68, 2236–2240. [Google Scholar] [CrossRef]
- Jeannot, M.A.; Cantwell, F.F. Mass Transfer Characteristics of Solvent Extraction into a Single Drop at the Tip of a Syringe Needle. Anal. Chem. 1997, 69, 235–239. [Google Scholar] [CrossRef]
- He, Y.; Lee, H.K. Liquid-Phase Microextraction in a Single Drop of Organic Solvent by Using a Conventional Microsyringe. Anal. Chem. 1997, 69, 4634–4640. [Google Scholar] [CrossRef]
- Ma, M.; Cantwell, F.F. Solvent Microextraction with Simultaneous Back-Extraction for Sample Cleanup and Preconcentration: Preconcentration into a Single Microdrop. Anal. Chem. 1999, 71, 388–393. [Google Scholar] [CrossRef]
- Baltussen, E.; Sandra, P.; David, F.; Cramers, C. Stir Bar Sorptive Extraction (SBSE), a Novel Extraction Technique for Aqueous Samples: Theory and Principles. J. Microcolumn Sep. 1999, 11, 737–747. [Google Scholar] [CrossRef]
- Rezaee, M.; Assadi, Y.; Milani Hosseini, M.-R.; Aghaee, E.; Ahmadi, F.; Berijani, S. Determination of Organic Compounds in Water Using Dispersive Liquid-Liquid Microextraction. J. Chromatogr. A 2006, 1116, 1–9. [Google Scholar] [CrossRef]
- Tsai, W.-H.; Huang, T.-C.; Huang, J.-J.; Hsue, Y.-H.; Chuang, H.-Y. Dispersive Solid-Phase Microextraction Method for Sample Extraction in the Analysis of Four Tetracyclines in Water and Milk Samples by High-Performance Liquid Chromatography with Diode-Array Detection. J. Chromatogr. A 2009, 1216, 2263–2269. [Google Scholar] [CrossRef] [PubMed]
- Shen, G.; Lee, H.K. Hollow Fiber-Protected Liquid-Phase Microextraction of Triazine Herbicides. Anal. Chem. 2002, 74, 648–654. [Google Scholar] [CrossRef] [PubMed]
- Basheer, C.; Alnedhary, A.A.; Rao, B.S.M.; Valliyaveettil, S.; Lee, H.K. Development and Application of Porous Membrane-Protected Carbon Nanotube Micro-Solid-Phase Extraction Combined with Gas Chromatography/Mass Spectrometry. Anal. Chem. 2006, 78, 2853–2858. [Google Scholar] [CrossRef]
- Nazal, M.K.; Sajid, M.; Gijjapu, D.R. Membrane-Based Inverted Liquid–Liquid Extraction of Organochlorine Pesticides in Aqueous Samples: Evaluation, Merits, and Demerits. Chem. Pap. 2023, 1–11. [Google Scholar] [CrossRef]
- Basheer, C.; Lee, H.K. Hollow Fiber Membrane-Protected Solid-Phase Microextraction of Triazine Herbicides in Bovine Milk and Sewage Sludge Samples. J. Chromatogr. A 2004, 1047, 189–194. [Google Scholar] [CrossRef] [PubMed]
- Esrafili, A.; Baharfar, M.; Tajik, M.; Yamini, Y.; Ghambarian, M. Two-Phase Hollow Fiber Liquid-Phase Microextraction. TrAC-Trends Anal. Chem. 2018, 108, 314–322. [Google Scholar] [CrossRef]
- Gjelstad, A. Three-Phase Hollow Fiber Liquid-Phase Microextraction and Parallel Artificial Liquid Membrane Extraction. TrAC-Trends Anal. Chem. 2019, 113, 25–31. [Google Scholar] [CrossRef]
- Sajid, M. Porous Membrane Protected Micro-Solid-Phase Extraction: A Review of Features, Advancements and Applications. Anal. Chim. Acta 2017, 965, 36–53. [Google Scholar] [CrossRef]
- Sajid, M.; Woźniak, M.K.; Płotka-Wasylka, J. Ultrasound-Assisted Solvent Extraction of Porous Membrane Packed Solid Samples: A New Approach for Extraction of Target Analytes from Solid Samples. Microchem. J. 2019, 144, 117–123. [Google Scholar] [CrossRef]
- Sajid, M.; Alhooshani, K. Ultrasound-Assisted Solvent Extraction of Organochlorine Pesticides from Porous Membrane Packed Tea Samples Followed by GC–MS Analysis. Microchem. J. 2020, 152, 104464. [Google Scholar] [CrossRef]
- Kafilzadeh, F. Assessment of Organochlorine Pesticide Residues in Water, Sediments and Fish from Lake Tashk, Iran. Achiev. Life Sci. 2015, 9, 107–111. [Google Scholar] [CrossRef] [Green Version]
- Sajid, M.; Basheer, C.; Narasimhan, K.; Buhmeida, A.; Qahtani, A.; Al-ahwal, M.S. Persistent and Endocrine Disrupting Organic Pollutants: Advancements and Challenges in Analysis, Health Concerns and Clinical Correlates. Nat. Environ. Pollut. Technol. 2016, 15, 733–746. [Google Scholar]
- Zhao, L.; Lee, H.K. Application of Static Liquid-Phase Microextraction to the Analysis of Organochlorine Pesticides in Water. J. Chromatogr. A 2001, 919, 381–388. [Google Scholar] [CrossRef] [PubMed]
- Hassan, A.A.; Sajid, M.; Al Ghafly, H.; Alhooshani, K. Ionic Liquid-Based Membrane-Protected Micro-Solid-Phase Extraction of Organochlorine Pesticides in Environmental Water Samples. Microchem. J. 2020, 158, 105295. [Google Scholar] [CrossRef]
- Płotka-Wasylka, J. A New Tool for the Evaluation of the Analytical Procedure: Green Analytical Procedure Index. Talanta 2018, 181, 204–209. [Google Scholar] [CrossRef]
- Pena-Pereira, F.; Wojnowski, W.; Tobiszewski, M. AGREE—Analytical GREEnness Metric Approach and Software. Anal. Chem. 2020, 92, 10076–10082. [Google Scholar] [CrossRef] [PubMed]
- Nowak, P.M.; Kościelniak, P. What Color Is Your Method? Adaptation of the Rgb Additive Color Model to Analytical Method Evaluation. Anal. Chem. 2019, 91, 10343–10352. [Google Scholar] [CrossRef]
- Gałuszka, A.; Migaszewski, Z.M.; Konieczka, P.; Namieśnik, J. Analytical Eco-Scale for Assessing the Greenness of Analytical Procedures. TrAC-Trends Anal. Chem. 2012, 37, 61–72. [Google Scholar] [CrossRef]
- NEMI. Available online: https://www.nemi.gov/about/ (accessed on 21 February 2023).
- Sajid, M.; Płotka-Wasylka, J. Green Analytical Chemistry Metrics: A Review. Talanta 2022, 238, 123046. [Google Scholar] [CrossRef]
- Lakshmi HimaBindu, M.R.; Angala Parameswari, S.; Gopinath, C. A Review on GC-MS and Method Development and Validation. Int. J. Pharm. Qual. Assur. 2013, 4, 42–51. [Google Scholar]
- Asati, A.; Satyanarayana, G.N.V.; Srivastava, V.T.; Patel, D.K. Determination of Organochlorine Compounds in Fish Liver by Ultrasound-Assisted Dispersive Liquid–Liquid Microextraction Based on Solidification of Organic Droplet Coupled with Gas Chromatography-Electron Capture Detection. J. Chromatogr. A 2018, 1561, 20–27. [Google Scholar] [CrossRef] [PubMed]
- Manuelmolina-Ruiz, J.; Cieslik, E.; Cieslik, I.; Walkowska, I. Determination of Pesticide Residues in Fish Tissues by Modified QuEChERS Method and Dual-d-SPE Clean-up Coupled to Gas Chromatography–Mass Spectrometry. Environ. Sci. Pollut. Res. 2014, 22, 369–378. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Yu, X.; He, X.; Xie, D.; Fan, Y.; Peng, J. Simplified Pesticide Multiresidues Analysis in Fish by Low-Temperature Cleanup and Solid-Phase Extraction Coupled with Gas Chromatography/Mass Spectrometry. Food Chem. 2009, 113, 1297–1300. [Google Scholar] [CrossRef]
- Shrivas, K.; Wu, H.F. Ultrasonication Followed by Single-Drop Microextraction Combined with GC/MS for Rapid Determination of Organochlorine Pesticides from Fish. J. Sep. Sci. 2008, 31, 380–386. [Google Scholar] [CrossRef] [PubMed]
- Darko, G.; Akoto, O.; Oppong, C. Persistent Organochlorine Pesticide Residues in Fish, Sediments and Water from Lake Bosomtwi, Ghana. Chemosphere 2008, 72, 21–24. [Google Scholar] [CrossRef]







| Parameter | Initial Value | Range Investigated in Optimization Experiments | Optimum Value |
|---|---|---|---|
| Extraction solvent | - | n-hexane, DCM, n-hexane:DCM (1:1), chloroform, methanol, toluene | Toluene |
| Extraction solvent volume (mL) | 1.0 | 0.5–5.0 | 3.0 |
| Mass of sample (mg) | 250 | 125–1000 | 500 |
| Ultrasound intensity | Medium | Low, medium, high | Medium |
| Extraction time (min) | 30 | 15–75 | 60 |
| Analyte | Correlation Coefficient (R2) | Linear Range (ng/g) | LOD (ng/g) | LOQ (ng/g) | RSDs (%) 100 ng/g (n = 6) | |
|---|---|---|---|---|---|---|
| Intra-Day | Inter-Day | |||||
| Heptachlor | 0.9957 | 10–1000 | 2.9 | 9.7 | 5.8 | 6.0 |
| Aldrin | 0.9989 | 5–1000 | 3.4 | 10.1 | 8.9 | 9.1 |
| Heptachlor Epoxide | 0.9922 | 10–1000 | 3.3 | 10.5 | 10.0 | 10.3 |
| Trans-chlordane | 0.9950 | 5–1000 | 3.4 | 10.3 | 9.7 | 10.3 |
| Endosulfan I | 0.9952 | 10–1000 | 3.4 | 10.5 | 9.6 | 11.6 |
| Cis-chlordane | 0.9956 | 20–1000 | 5.8 | 19.7 | 8.9 | 9.5 |
| Dieldrin | 0.9995 | 5–1000 | 1.5 | 5.0 | 8.9 | 10.4 |
| 4,4′-DDE | 0.9979 | 10–1000 | 3.1 | 10.2 | 9.3 | 11.1 |
| Endrin | 0.9974 | 10–1000 | 2.9 | 9.9 | 5.6 | 7.7 |
| Endosulfan II | 0.9991 | 10–1000 | 3.2 | 10.7 | 10.8 | 11.9 |
| 4,4′-DDD | 0.9962 | 10–1000 | 3.3 | 10.1 | 8.3 | 8.5 |
| Endrin Aldehyde | 0.9988 | 10–1000 | 3.2 | 10.1 | 8.6 | 9.1 |
| Endosulfan Sulfate | 0.9967 | 20–1000 | 6.8 | 19.7 | 13.5 | 13.8 |
| 4,4′-DDT | 0.9989 | 10–1000 | 3.4 | 9.6 | 10.6 | 11.2 |
| Methoxychlor | 0.9982 | 5–1000 | 1.8 | 5.2 | 9.1 | 10.3 |
| Analyte | Spike Level 50 ng/g | Spike Level 100 ng/g | Spike Level 250 ng/g | |||
|---|---|---|---|---|---|---|
| Relative Recovery (%) | RSDs (%) | Relative Recovery (%) | RSDs (%) | Relative Recovery (%) | RSDs (%) | |
| Heptachlor | 96.6 | 5.1 | 98.4 | 4.7 | 93.8 | 3.2 |
| Aldrin | 95.1 | 3.7 | 96.3 | 5.0 | 92.2 | 6.0 |
| Heptachlor Epoxide | 92.5 | 4.9 | 96.2 | 2.9 | 94.6 | 3.2 |
| Trans-chlordane | 89.7 | 5.4 | 91.5 | 4.4 | 94.6 | 4.9 |
| Endosulfan I | 89.5 | 4.6 | 96.8 | 4.9 | 94.5 | 6.1 |
| Cis-chlordane | 94.1 | 4.2 | 91.9 | 3.1 | 98.1 | 4.6 |
| Dieldrin | 96.6 | 3.8 | 97.4 | 2.0 | 89.2 | 2.9 |
| 4,4′-DDE | 85.7 | 4.8 | 92.7 | 5.1 | 87.9 | 5.3 |
| Endrin | 88.2 | 4.3 | 91.6 | 4.5 | 89.8 | 3.9 |
| Endosulfan II | 98.4 | 2.9 | 96.1 | 5.1 | 90.0 | 6.0 |
| 4,4′-DDD | 92.1 | 4.1 | 95.4 | 4.6 | 91.5 | 4.5 |
| Endrin Aldehyde | 99.1 | 3.6 | 98.2 | 3.3 | 104.2 | 4.1 |
| Endosulfan Sulfate | 87.3 | 3.2 | 88.5 | 5.1 | 93.4 | 3.8 |
| 4,4′-DDT | 96.1 | 1.8 | 100.3 | 4.0 | 100.1 | 6.8 |
| Methoxychlor | 88.2 | 2.2 | 86.1 | 2.5 | 88.6 | 3.5 |
| Method | Solvents Employed and Volume | Linear Range | LOD (ng/g) | RSDs (%) | Recoveries (%) | Ref. |
|---|---|---|---|---|---|---|
| US-DLLME-SFO-GC-μECD | Acetone (5 mL) and 1-Undecanol (24 μL) | 1–500 | 1.06–3.84 | <6.3 | 88.5–108.4 | [42] |
| QuEChERS-d-SPE-GC-MS | Acetonitrile (10 mL), chloroform (2 mL) | 3–200 ng/mL | 1.0–3.0 | <10 | 70–120 | [43] |
| LTC-SPE-GC–MS | Acetonitrile (30 mL), acetonitrile–toluene (3:1) (39 mL), n-hexane (1 mL) | - | 0.5–20 | <13.5 | 78.7–113.7 | [44] |
| U-SDME-GC-MS | Methanol (1 mL), toluene (1 µL) | 10–1000 | 0.5 | 9.4–10 | 82.1–95.3 | [45] |
| Soxhlet Extraction-µC-GC-ECD | Acetone: n-hexane (20:80 v/v) (150 mL), n-hexane (13.5 mL) | - | 0.6– 3.0 ng/L | 5.0–13 | 78–95 | [46] |
| UAE of membrane packed fish samples-GC-MS | Toluene (3 mL) | 5–1000, 10–1000 and 20–1000 | 1.5–6.8 | ≤13.8 | 87.3–104.2 | This work |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sajid, M.; Alhooshani, K. Extraction of Organochlorine Pesticides from Porous Membrane Packed Dried Fish Samples: Method Development and Greenness Evaluation. Separations 2023, 10, 233. https://doi.org/10.3390/separations10040233
Sajid M, Alhooshani K. Extraction of Organochlorine Pesticides from Porous Membrane Packed Dried Fish Samples: Method Development and Greenness Evaluation. Separations. 2023; 10(4):233. https://doi.org/10.3390/separations10040233
Chicago/Turabian StyleSajid, Muhammad, and Khalid Alhooshani. 2023. "Extraction of Organochlorine Pesticides from Porous Membrane Packed Dried Fish Samples: Method Development and Greenness Evaluation" Separations 10, no. 4: 233. https://doi.org/10.3390/separations10040233