
A Validated, Stability-Indicating, Eco-Friendly HPTLC Method for the Determination of Cinnarizine

 , , , , ,
, , , , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Equipment and Analytical Conditions
2.3. Calibration Curve for CIN
2.4. Sample Preparation for the Assay of CIN in Commercial Tablets
2.5. Validation Studies
2.6. Forced Degradation/Selectivity Investigations
2.7. Application of the Existing Approach in the Measurement of CIN in Commercial Tablets
2.8. Greenness Measurement
3. Results and Discussion
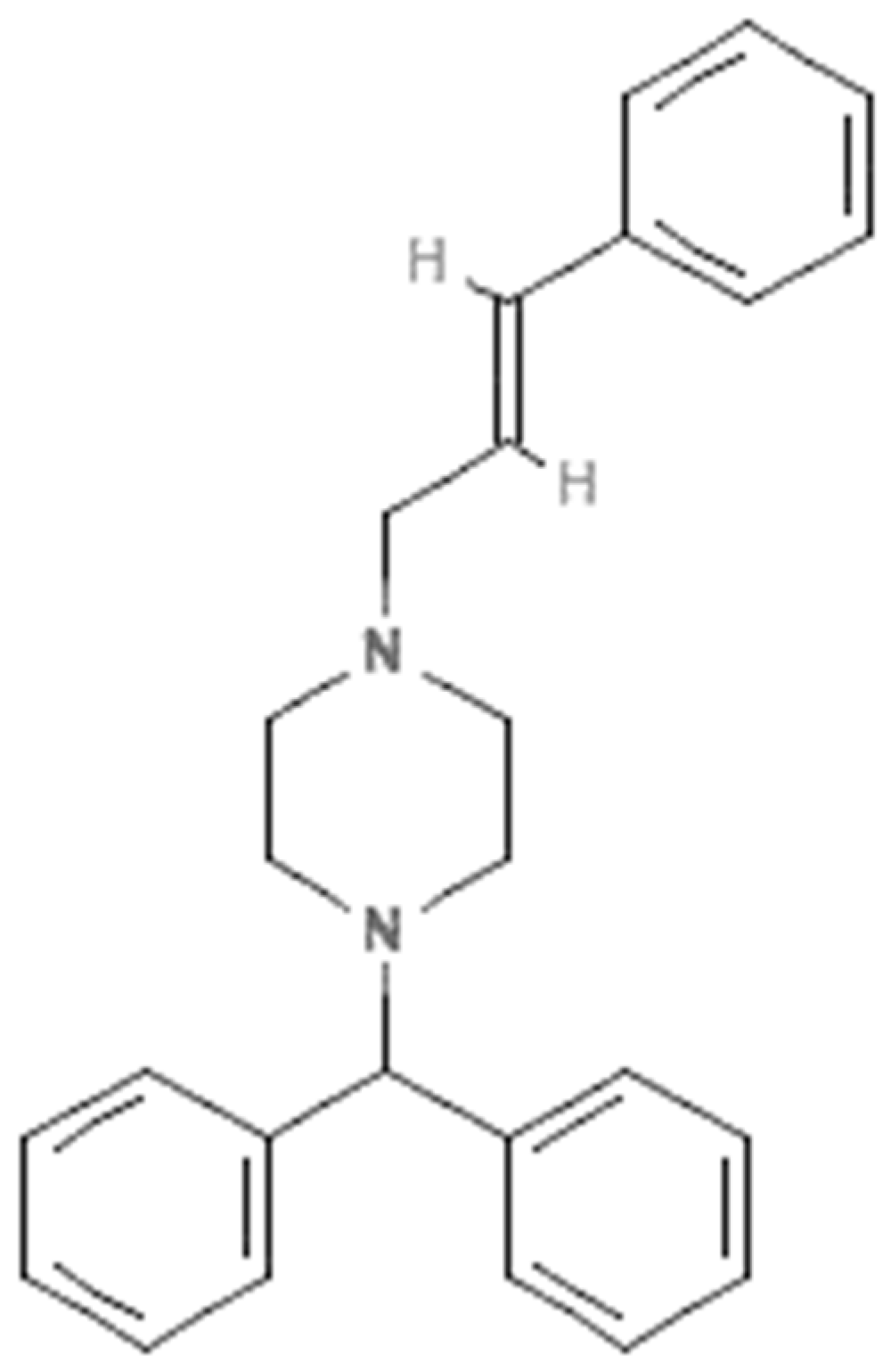
3.1. Method Development
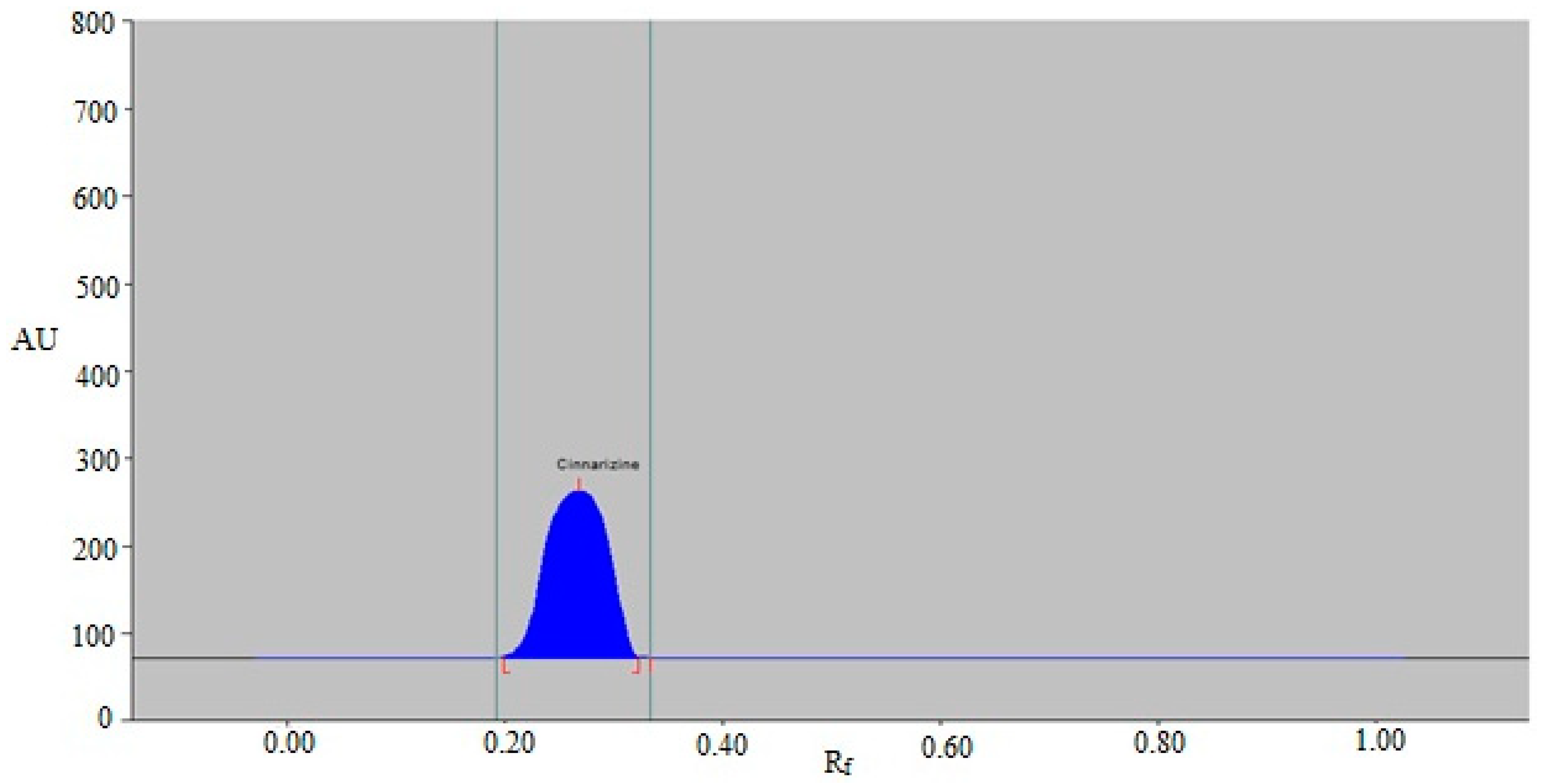
3.2. Analytical Method Validation
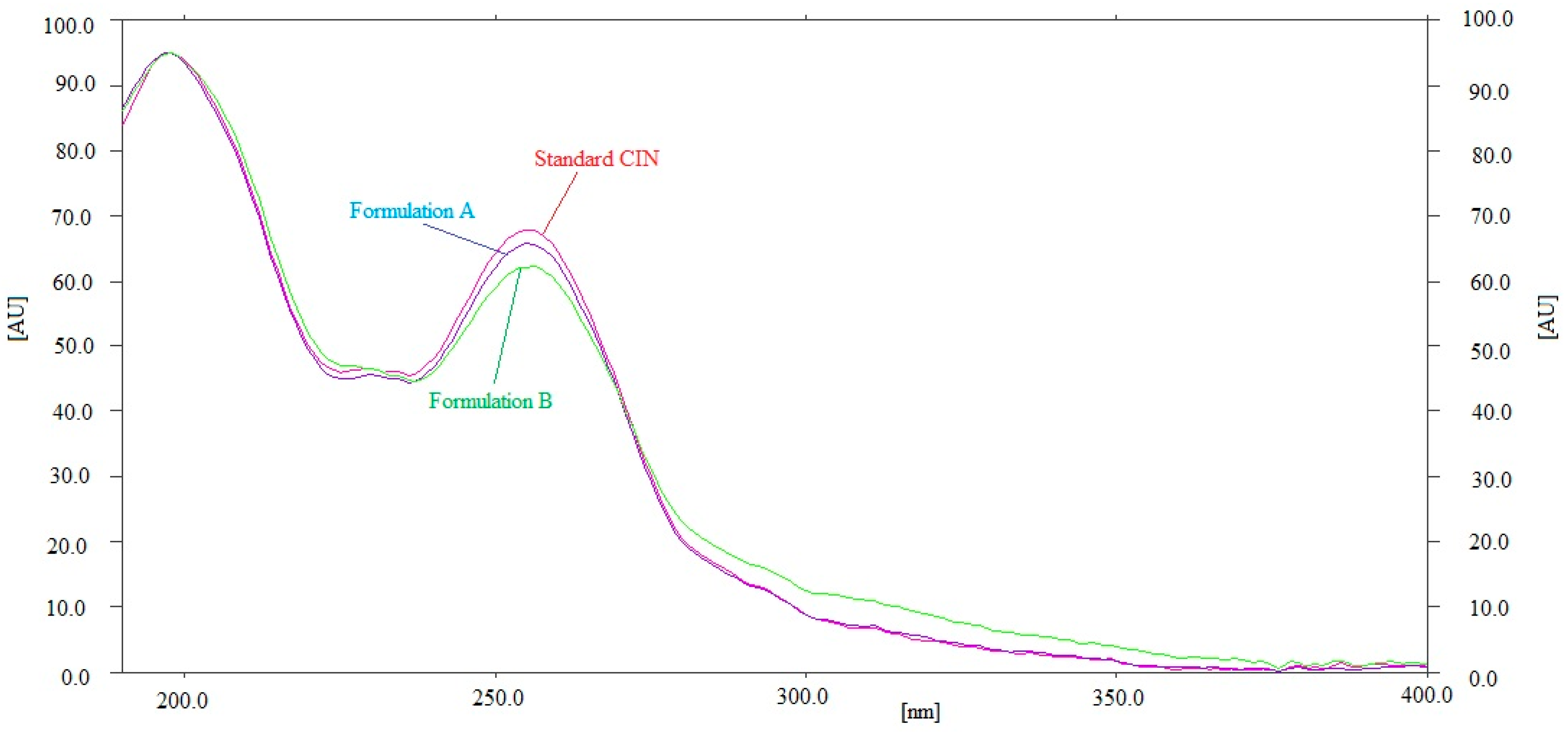
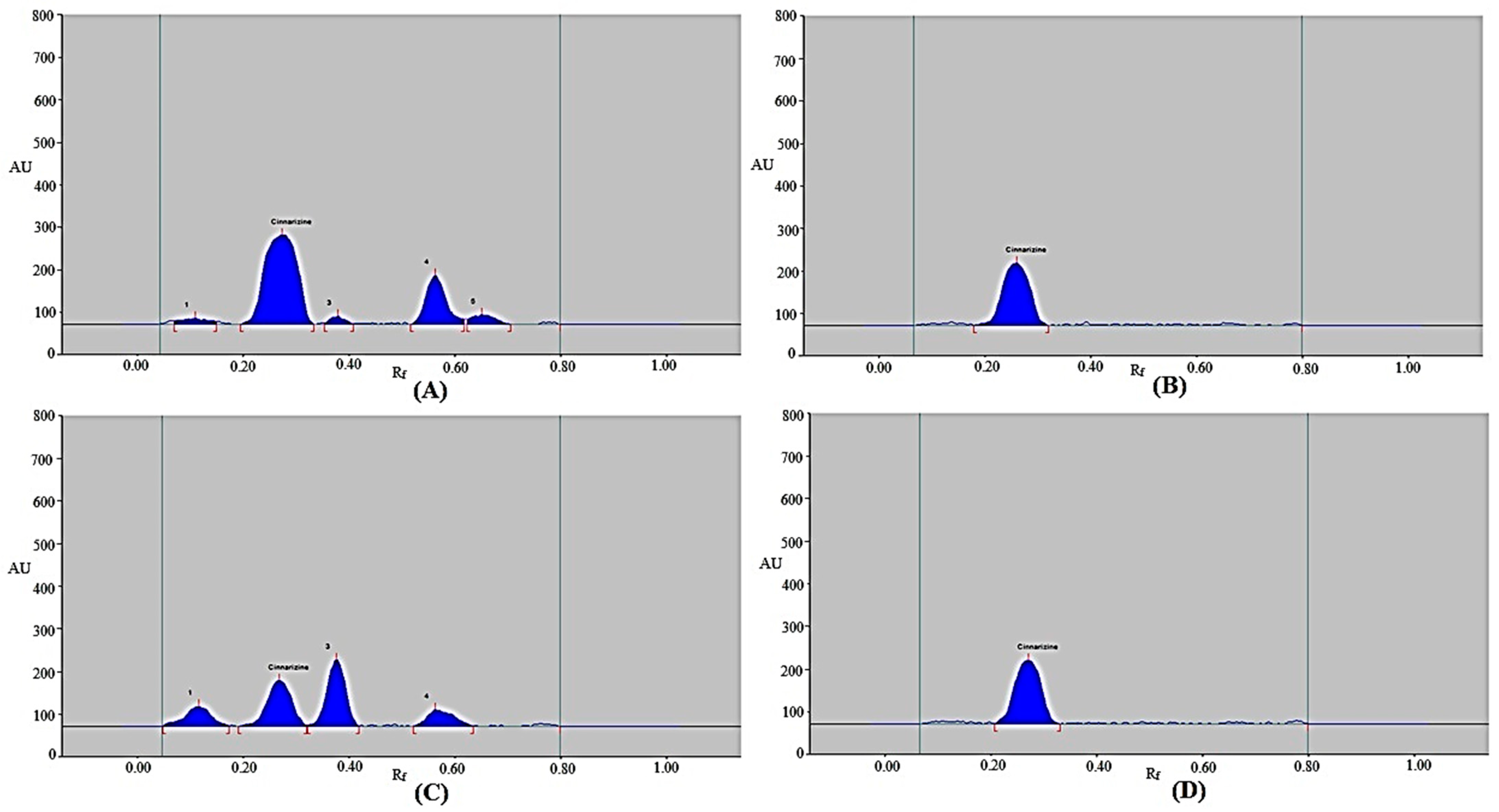
3.3. Selectivity/Forced Degradation Investigations
3.4. Application of Existing Approach in the Measurement of CIN in Commercial Tablets
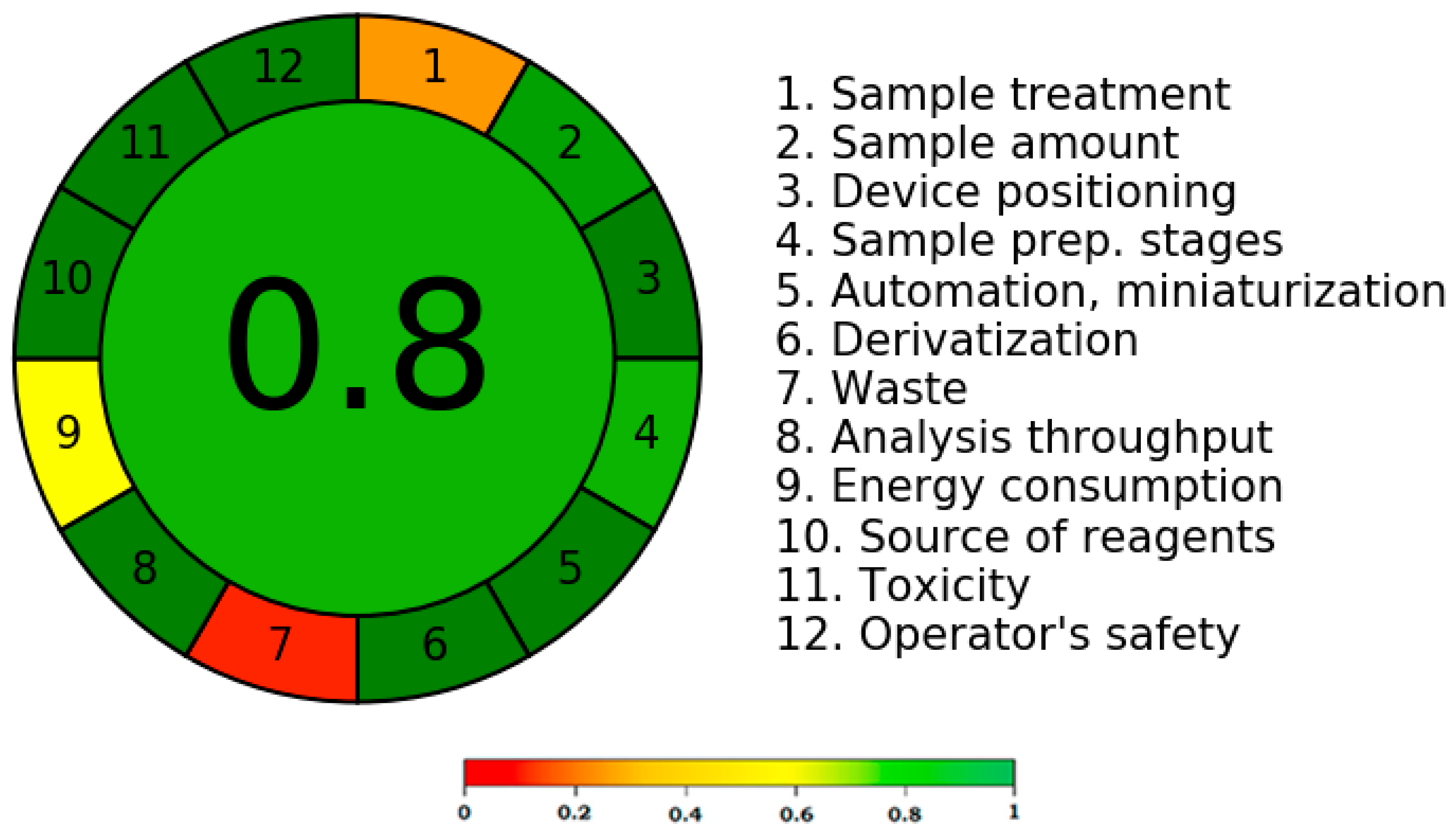
3.5. Greenness Measurement
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Raghuvanshi, S.; Pathak, K. Recent advances in drug delivery systems and therapeutics of cinnarizine: A poorly water soluble drug with absorption window in stomach. J. Drug Deliv. 2014, 2014, E479246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, S.; Chen, H.; Lin, X.; Tang, X. Pharmacokinetics, tissue distribution and safety of cinnarizine delivered in lipid emulsion. Int. J. Pharm. 2010, 383, 264–270. [Google Scholar] [CrossRef] [PubMed]
- Lucertini, M.; Mirante, N.; Casagrande, M.; Trivelloni, P.; Lugli, V. The effect of cinnarizine and cocculus indicus on simulator sickness. Physiol. Behav. 2007, 91, 180–190. [Google Scholar] [CrossRef] [PubMed]
- Doweck, I.; Gordon, C.R.; Spitzer, O.; Melamed, Y.; Shupak, A. Effect of cinnarizine in the prevention of seasickness. Aviat. Space Environ. Med. 1994, 65, 606–609. [Google Scholar]
- Gordon, C.R.; Gonen, A.; Nachum, Z.; Dowech, I.; Spitzer, O.; Shupak, A. The effects of dimenhydrinate, cinnarizine, and transdermal scopolamine on performance. J. Psychopharmacol. 2001, 15, 167–172. [Google Scholar] [CrossRef]
- Shakeel, F.; Kazi, M.; Alanazi, F.K.; Alam, P. Solubility of cinnarizine in (Transcutol + water) mixtures: Determination, Hansen solubility parameters, correlation, and thermodynamics. Molecules 2021, 26, 7052. [Google Scholar] [CrossRef]
- Amidon, G.L.; Lennernas, H.; Shah, V.P.; Crison, J.R. A theoretical basis for a biopharmaceutic drug classification-the correlation of in-vitro drug product dissolution and in-vivo bioavailability. Pharm. Res. 1995, 12, 413–420. [Google Scholar] [CrossRef] [Green Version]
- Pedersen, P.B.; Berthelsen, R.; Rades, T.; Jorgensen, S.A.; Vilmann, P.; Bar-Shalom, D.; Baldursdottir, S.; Mullertz, A. Physico-chemical characterization of aspirated and simulated human gastric fluids to study their influence on the intrinsic dissolution rate of cinnarizine. Int. J. Pharm. 2022, 622, E121856. [Google Scholar] [CrossRef]
- Albertini, B.; Bertoni, S.; Sangiorgi, S.; Nucci, G.; Resources, N.P.; Mezzina, E. NaDES as a green technological approach for the solubility improvement of BCS class II APIs: An insight into the molecular interactions. Int. J. Pharm. 2023, 634, E122696. [Google Scholar] [CrossRef]
- Abdine, H.; Belal, F.; Zoman, N. Simple spectrophotometric determination of cinnarizine in its dosage forms. Farmaco 2002, 57, 267–271. [Google Scholar] [CrossRef]
- Salem, M.Y.; El-Bardicy, M.G.; El-Tarras, M.F.; El-Zanfally, E.S. Simultaneous determination of domperidone maleate and cinnarizine in a binary mixture using derivative ratio spectrophotometry and classical least squares calibration. J. Pharm. Biomed. Anal. 2002, 30, 21–33. [Google Scholar] [CrossRef] [PubMed]
- Salem, M.Y.; El-Zanfally, E.S.; El-Tarras, M.F.; El-Bardicy, M.G. Simultaneous determination of domperidone and cinnarizine in a binary mixture using derivative spectrophotometry, partial least squares and principle component regression calibration. Anal. Bioanal. Chem. 2003, 375, 211–216. [Google Scholar] [CrossRef] [PubMed]
- Devagondanahalli, M.H.; Shaikh, S.M.T.; Jaldappagari, S.; Ramanaboyina, S.K.; Kasalanti, H. Determination of cinnarizine in pure and pharmaceutical formulations. J. Chin. Chem. Soc. 2007, 54, 63–68. [Google Scholar] [CrossRef]
- Abdelrahman, M.M. Simultaneous determination of cinnarizine and domperidone by area under curve and dual wavelength spectrophotometric methods. Spectrochim. Acta Part A 2013, 113, 291–296. [Google Scholar] [CrossRef]
- Lamie, N.T.; Yehia, A.M. Development of normalized spectra manipulating spectrophotometric methods for simultaneous determination of dimenhydrinate and cinnarizine binary mixture. Spectrochim. Acta Part A 2015, 150, 142–150. [Google Scholar] [CrossRef]
- Al-Ghani, A.M.; Thabet, A.A.M. Validated spectrophotometric methods for the estimation of cinnarizine in binary mixture with paracetamol in bulk and tablets. Asian J. Pharm. Clin. Res. 2021, 14, 161–166. [Google Scholar] [CrossRef]
- Heda, A.A.; Sonawane, A.R.; Naranje, G.H.; Puranik, P.K. A rapid determination of cinnarizine in bulk and pharmaceutical dosage form by LC. E.-J. Chem. 2010, 7, 1080–1084. [Google Scholar] [CrossRef] [Green Version]
- El-Houssini, O.M.; Zawilla, N.H.; Mohammad, M.A. Development and validation of RP-LC method for the determination of cinnarizine/piracetam and cinnarizine/heptaminol acefyllinate in presence of cinnarizine reported degradation products. Anal. Chem. Insights 2013, 8, 99–106. [Google Scholar] [CrossRef] [Green Version]
- Naga Sirisha, M.; Shanta Kumari, A. Validated RP-HPLC method for simultaneous estimation of cinnarizine and domperidone in bulk and pharmaceutical dosage form. J. Pharm. Scient. Innov. 2013, 2, 46–50. [Google Scholar]
- El-Adl, S.M.; El-Sadek, M.E.; Hasan, M.H. Exploring novel isocractic HPLC method for quantitative determination of cinnarizine and piracetam in their capsule preparations. J. Appl. Pharm. 2016, 8, E1000225. [Google Scholar]
- Edrees, F.H.; Saad, A.S.; Alsaadi, M.T.; Amin, N.H.; Abdelwahab, N.S. Experimentally designed chromatographic method for the simultaneous analysis of dimenhydrinate, cinnarizine and their toxic impurities. RSC Adv. 2021, 11, 1450–1460. [Google Scholar] [CrossRef] [PubMed]
- Hundt, H.K.L.; Brown, L.W.; Clark, E.C. Determination of cinnarizine in plasma by high-performance liquid chromatography. J. Chromatogr. B 1980, 183, 378–382. [Google Scholar] [CrossRef]
- Hundt, H.K.L. Determination of cinnarizine in plasma by high performance liquid chromatography. J. Chromatogr. B 1982, 232, 465. [Google Scholar] [CrossRef] [PubMed]
- Nowacka-Krukowska, H.; Rakowska, M.; Neubart, K.; Kobylinska, M. High-performance liquid chromatographic assay for cinnarizine in human plasma. Acta Pol. Pharm. 2007, 63, 407–411. [Google Scholar]
- Mullangi, S.; Ravindhranath, K.; Yarala, M.R.; Panchakarla, R.K. A sensitive LC-MS/MS method for the determination of genotoxic impurities in cinnarizine. Anal. Pharm. Franc. 2023, 81, 74–82. [Google Scholar] [CrossRef] [PubMed]
- Lamie, N.T.; Monir, H.H. Simultaneous determination of cinnarizine and dimenhydrinate in binary mixture using chromatographic methods. J. Chromatogr. Sci. 2016, 54, 36–42. [Google Scholar] [CrossRef] [Green Version]
- Ahmed, A.B.; Abdelwahab, N.S.; Abdelrahman, M.M.; Salama, F.M. Simultaneous determination of dimenhydrinate, cinnarizine and cinnarizine impurity by TLC and HPLC chromatographic methods. Bull. Fac. Pharm. Cairo Univ. 2017, 55, 163–169. [Google Scholar] [CrossRef]
- Mahrouse, M.A.; El-Zaher, A.A.; Al-Ghani, A.M. Validated chromatographic methods for simultaneous estimation of cinnarizine in binary mixture with domperidone and paracetamol in tablets. Curr. Pharm. Anal. 2019, 15, 429–438. [Google Scholar] [CrossRef]
- Hassan, S.S.M.; Elmosallamy, M.A.F.; Abbas, A.B. LC and TLC determination of cinnarizine in pharmaceutical preparations and serum. J. Pharm. Biomed. Anal. 2002, 28, 711–719. [Google Scholar] [CrossRef]
- El-Houssini, O.M.; Mohammad, M.A. Versatile TLC-densitometric methods for the synchronous estimation of cinnarizine and acefylline heptaminol in the presence of potential impurity and their reported degradation products. J. Chromatogr. Sci. 2022, 60, 832–839. [Google Scholar] [CrossRef]
- Mhaske, D.K.; Kumbhar, A.S. Development and validation of rapid, timesaving, and cost-effective UHPLC method for simultaneous quantification of cinnarizine, its five specified impurities, two degradation products and two antioxidants. Anal. Chem. Lett. 2022, 12, 488–504. [Google Scholar] [CrossRef]
- Trivedi, J.U.; Ghalsasi, P.; Ganguly, S.; Mary, S.J.J.; James, C. Raman spectroscopic study of cinnamyl-1 diphenylmethyl-4 piperazine (cinnarizine) at high pressure. J. Mol. Str. 2022, 1253, E132214. [Google Scholar] [CrossRef]
- El-Sayed, G.O.; Yasin, S.A.; El Badawy, A.A. Voltammetric behavior and determination of cinnarizine in pharmaceutical formulations and serum. Anal. Lett. 2008, 41, 3021–3033. [Google Scholar] [CrossRef]
- Hedge, R.N.; Hosamani, R.R.; Nandibewoor, S.T. Voltammetric oxidation and determination of cinnarizine at glassy carbon electrode modified with multi-walled carbon nanotubes. Coll. Surf. B 2009, 72, 259–265. [Google Scholar]
- Walash, M.I.; Belal, F.; El-Enany, N.; Abdelal, A.A. Second-derivative synchronous fluorometric method for the simultaneous determination of cinnarizine and domperidone in pharmaceutical preparations. Application to biological fluids. J. Fluoresc. 2008, 18, 61–74. [Google Scholar] [CrossRef] [PubMed]
- Walash, M.I.; Belal, F.; El-Enany, N.; Abdelal, A. Second-derivative synchronous fluorescence spectroscopy for the simultaneous determination of cinnarizine and nicergoline in pharmaceutical preparations. J. AOAC Int. 2008, 91, 349–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Zhang, Y.; Liu, X.; Xu, X.; Li, Y.; Zhang, T. Supercritical fluid chromatography tandem mass spectrometry employed with evaporation-free liquid–liquid extraction for the rapid analysis of cinnarizine in rat plasma. J. Sep. Sci. 2021, 45, 968–975. [Google Scholar] [CrossRef]
- Woestenborghs, R.; Michielsen, L.; Lorreyne, W.; Heykants, J. Sensitive gas chromatographic method for the determination of cinnarizine and flunarizine in biological samples. J. Chromatogr. B 1982, 232, 85–91. [Google Scholar] [CrossRef]
- Abdelal, A.A.; Kitagawa, S.; Ohtani, H.; El-Enany, N.; Belal, F.; Walash, M.I. Method development and validation for the simultaneous determination of cinnarizine and co-formulated drugs in pharmaceutical preparations by capillary electrophoresis. J. Pharm. Biomed. Anal. 2008, 46, 491–497. [Google Scholar] [CrossRef]
- Tawakkol, S.M.; El-Zeiny, M.B.; Hemdan, A. Full spectrum and selected spectrum based chemometric methods for the simultaneous determination of cinnarizine and dimenhydrinate in laboratory prepared mixtures and pharmaceutical dosage form. Spectrochim. Acta A 2017, 173, 892–896. [Google Scholar] [CrossRef]
- Abdelrahman, M.M.; Abdelwahab, N.S.; Hegazy, M.A.; Fares, M.Y.; El-Sayed, G.M. Determination of the abused intravenously administered madness drops (tropicamide) by liquid chromatography in rat plasma; an application to pharmacokinetic study and greenness profile assessment. Microchem. J. 2020, 159, E105582. [Google Scholar] [CrossRef]
- Duan, X.; Liu, X.; Dong, Y.; Yang, J.; Zhang, J.; He, S.; Yang, F.; Wang, Z.; Dong, Y. A green HPLC method for determination of nine sulfonamides in milk and beef, and its greenness assessment with analytical eco-scale and greenness profile. J. AOAC Int. 2020, 103, 1181–1189. [Google Scholar] [CrossRef] [PubMed]
- Pena-Pereira, F.; Wojnowski, W.; Tobiszewski, M. AGREE-Analytical GREEnness metric approach and software. Anal. Chem. 2020, 92, 10076–10082. [Google Scholar] [CrossRef]
- Alam, P.; Salem-Bekhit, M.M.; Al-Joufi, F.A.; Alqarni, M.H.; Shakeel, F. Quantitative analysis of cabozantinib in pharmaceutical dosage forms using green RP-HPTLC and green NP-HPTLC methods: A comparative evaluation. Sus. Chem. Pharm. 2021, 21, E100413. [Google Scholar] [CrossRef]
- Foudah, A.I.; Shakeel, F.; Alqarni, M.H.; Alam, P. A rapid and sensitive stability-indicating green RP-HPTLC method for the quantitation of flibanserin compared to green NP-HPTLC method: Validation studies and greenness assessment. Microchem. J. 2021, 164, E105960. [Google Scholar] [CrossRef]
- International Conference on Harmonization (ICH). Q2 (R1): Validation of Analytical Procedures–Text and Methodology. Geneva, Switzerland, 2005. Available online: https://database.ich.org/sites/default/files/Q2%28R1%29%20Guideline.pdf (accessed on 1 February 2023).
- Alam, P.; Shakeel, F.; Ali, A.; Alqarni, M.H.; Foudah, A.I.; Aljarba, T.M.; Alkholifi, F.K.; Alshehri, S.; Ghoneim, M.M.; Ali, A. Simultaneous determination of caffeine and paracetamol in commercial formulations using greener normal-phase and reversed-phase HPTLC methods: A contrast of validation parameters. Molecules 2022, 27, 405. [Google Scholar] [CrossRef] [PubMed]
- Haq, N.; Iqbal, M.; Alanazi, F.K.; Alsarra, I.A.; Shakeel, F. Applying green analytical chemistry for rapid analysis of drugs: Adding health to pharmaceutical industry. Arabian J. Chem. 2017, 10, S777–S785. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Eco-Friendly Mobile Phase | As | N m−1 | Rf |
|---|---|---|---|
| EtOH-H2O (30:70 v/v) | 1.31 ± 0.05 | 1548 ± 1.78 | 0.33 ± 0.02 |
| EtOH-H2O (40:60 v/v) | 1.30 ± 0.04 | 1741 ± 1.85 | 0.32 ± 0.03 |
| EtOH-H2O (50:50 v/v) | 1.29 ± 0.03 | 1904 ± 1.92 | 0.31 ± 0.03 |
| EtOH-H2O (60:40, v/v) | 1.28 ± 0.04 | 2274 ± 1.98 | 0.30 ± 0.02 |
| EtOH-H2O (70:30 v/v) | 1.27 ± 0.03 | 2841 ± 2.45 | 0.29 ± 0.02 |
| EtOH-H2O (80:20 v/v) | 1.26 ± 0.02 | 3452 ± 2.84 | 0.28 ± 0.01 |
| EtOH-H2O (90:10 v/v) | 1.10 ± 0.03 | 4523 ± 3.78 | 0.27 ± 0.01 |
| Parameters | Value |
|---|---|
| Linearity range (ng band−1) | 50–800 |
| Regression equation | y = 12.224x + 507.44 |
| R2 | 0.9976 |
| R | 0.9987 |
| SE of slope | 0.27 |
| SE of intercept | 1.73 |
| 95% CI of slope | 11.04–13.40 |
| 95% CI of intercept | 499.99–514.88 |
| LOD ± SD (ng band−1) | 16.81 ± 0.09 |
| LOQ ± SD (ng band−1) | 50.43 ± 0.27 |
| Concentration (ng band−1) | Concentration Found (ng band−1) ± SD | Recovery (%) | CV (%) |
|---|---|---|---|
| Intra-day accuracy | |||
| 300 | 302.12 ± 2.64 | 100.70 | 0.87 |
| 400 | 405.16 ± 3.36 | 101.29 | 0.82 |
| 500 | 496.21 ± 4.02 | 99.24 | 0.81 |
| Inter-day accuracy | |||
| 300 | 298.41 ± 2.66 | 99.47 | 0.89 |
| 400 | 396.31 ± 3.34 | 99.07 | 0.84 |
| 500 | 506.31 ± 4.17 | 101.26 | 0.82 |
| Concentration (ng band−1) | Intra-Day Precision | Inter-Day Precision | ||||
|---|---|---|---|---|---|---|
| Concentration (ng band−1) ± SD | SE | CV (%) | Concentration (ng band−1) ± SD | SE | CV (%) | |
| 300 | 303.16 ± 2.77 | 1.13 | 0.91 | 305.21 ± 2.90 | 1.18 | 0.95 |
| 400 | 397.41 ± 3.41 | 1.39 | 0.85 | 407.24 ± 3.56 | 1.45 | 0.87 |
| 500 | 504.61 ± 4.06 | 1.65 | 0.80 | 494.63 ± 4.21 | 1.71 | 0.85 |
| Concentration (ng band−1) | Eco-Friendly Mobile Phase (EtOH-H2O) | Results | ||||
|---|---|---|---|---|---|---|
| Original | Used | Level | Concentration (ng band−1) ± SD | CV (%) | Rf | |
| 92:8 | +2.0 | 393.32 ± 3.65 | 0.92 | 0.26 | ||
| 400 | 90:10 | 90:10 | 0.0 | 398.21 ± 3.91 | 0.98 | 0.27 |
| 88:12 | −2.0 | 407.81 ± 4.17 | 1.02 | 0.28 | ||
| Stress Condition | Number of Degradation Products (Rf) | CIN Rf | CIN Remained (ng band−1) | CIN Recovered (%) |
|---|---|---|---|---|
| 1 M HCl | 4 (0.11, 0.38, 0.56, 0.65) | 0.27 | 265.60 | 66.40 ± 1.18 |
| 1 M NaOH | 0 | 0.26 | 400.00 | 100.00 ± 0.00 |
| 30% H2O2 | 3 (0.12, 0.38, 0.56) | 0.27 | 130.72 | 32.68 ± 0.58 |
| Thermal | 0 | 0.27 | 400.00 | 100 ± 0.00 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alqarni, M.H.; Shakeel, F.; Foudah, A.I.; Aljarba, T.M.; Mahdi, W.A.; Bar, F.M.A.; Alshehri, S.; Alam, P. A Validated, Stability-Indicating, Eco-Friendly HPTLC Method for the Determination of Cinnarizine. Separations 2023, 10, 138. https://doi.org/10.3390/separations10020138
Alqarni MH, Shakeel F, Foudah AI, Aljarba TM, Mahdi WA, Bar FMA, Alshehri S, Alam P. A Validated, Stability-Indicating, Eco-Friendly HPTLC Method for the Determination of Cinnarizine. Separations. 2023; 10(2):138. https://doi.org/10.3390/separations10020138
Chicago/Turabian StyleAlqarni, Mohammed H., Faiyaz Shakeel, Ahmed I. Foudah, Tariq M. Aljarba, Wael A. Mahdi, Fatma M. Abdel Bar, Sultan Alshehri, and Prawez Alam. 2023. "A Validated, Stability-Indicating, Eco-Friendly HPTLC Method for the Determination of Cinnarizine" Separations 10, no. 2: 138. https://doi.org/10.3390/separations10020138