
Diagnosing Cutaneous Melanocytic Tumors in the Molecular Era: Updates and Review of Literature


Abstract
:1. Introduction
2. Background of the Genomic Landscape
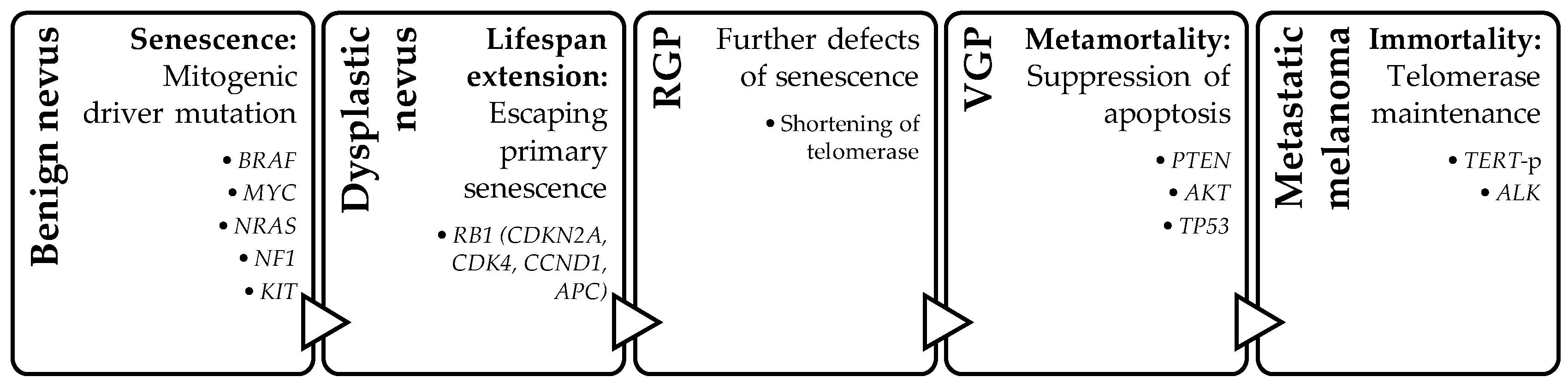
Four-Step Model
3. Genetic Change: Dysregulation of Three Main Oncogenic Signaling Pathways
3.1. MAPK Pathway
3.2. PI3K-AKT Pathway
3.3. Wnt/β-Catenin Pathway
4. Epigenetic Changes: DNA Methylation, Histone Modification, and MicroRNA Dysregulation
4.1. DNA Methylation Changes
4.2. Histone Modifications
4.3. MicroRNA Dysregulation
5. Practical Molecular Knowledge for a Diagnostic Surgical Pathologist
5.1. IHC for Assessment of Molecular Alteration
5.1.1. Genomic Events That Can Be Assessed Using IHC
VE1
NRASQ16R
ALK, ROS1, and Pan-TRK
β-Catenin and LEF1
BAP1
PRKAR1A
P16
5.1.2. Selected Melanocytic Lesions
Spitz Family
Deep Penetrating Nevus
5.2. Molecular Tests
5.2.1. Genomic Copy Number Assessment
CGH/SNP/MIP
Fluorescence in Situ Hybridization (FISH)
5.2.2. Gene Expression Profiling
5.2.3. Mutation Analysis: Telomerase Reverse Transcriptase Promoter
5.2.4. Imaging Mass Spectrometry
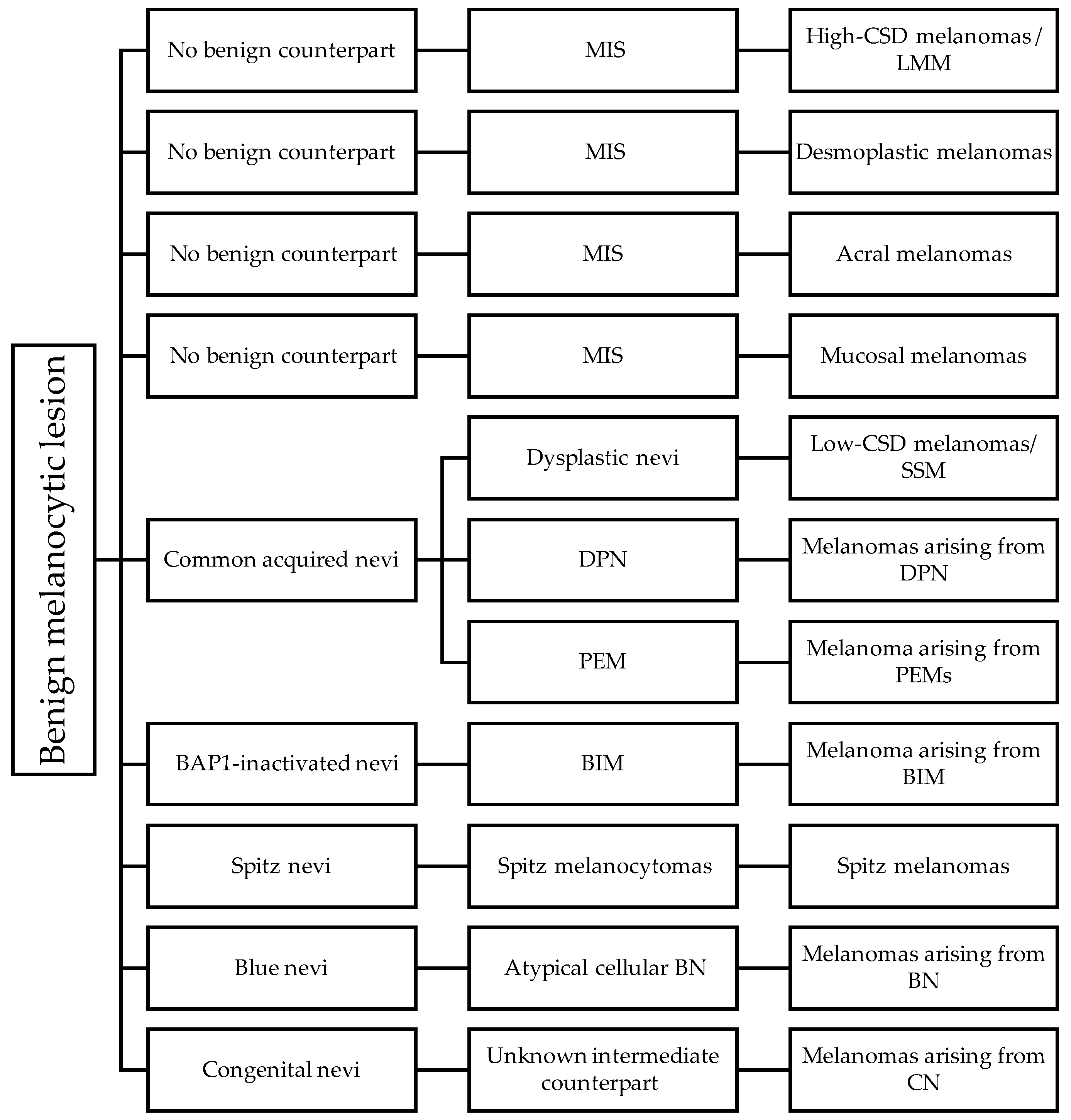
6. The Pathways of Cutaneous Melanomas
6.1. Pathway 1: Low-CSD/Superficial Spreading Melanoma
6.2. Pathway 2: High-CSD/Lentigo Maligna Melanoma
6.3. Pathway 3: Desmoplastic Melanoma
6.4. Pathway 4: Spitz Melanoma
6.5. Pathway 5: Acral Melanoma
6.6. Pathway 6: Mucosal Melanoma
6.7. Pathway 7: Melanoma Arising from Congenital Nevus
6.8. Pathway 8: Melanoma Arising from Blue Nevus
7. Discussion
8. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Hayward, N.K.; Wilmott, J.S.; Waddell, N.; Johansson, P.A.; Field, M.A.; Nones, K.; Patch, A.M.; Kakavand, H.; Alexandrov, L.B.; Burke, H.; et al. Whole-genome landscapes of major melanoma subtypes. Nature 2017, 545, 175–180. [Google Scholar] [CrossRef]
- Bennett, D.C. Genetics of melanoma progression: The rise and fall of cell senescence. Pigment Cell Melanoma Res. 2016, 29, 122–140. [Google Scholar] [CrossRef]
- Sullivan, R.; Flaherty, K. MAP kinase signaling and inhibition in melanoma. Oncogene 2013, 32, 2373–2379. [Google Scholar] [CrossRef] [PubMed]
- Guo, W.; Wang, H.; Li, C. Signal pathways of melanoma and targeted therapy. Signal Transduct. Target. Ther. 2021, 6, 424. [Google Scholar] [CrossRef]
- Platz, A.; Egyhazi, S.; Ringborg, U.; Hansson, J. Human cutaneous melanoma; a review of NRAS and BRAF mutation frequencies in relation to histogenetic subclass and body site. Mol. Oncol. 2008, 1, 395–405. [Google Scholar] [CrossRef]
- Hodis, E.; Watson, I.R.; Kryukov, G.V.; Arold, S.T.; Imielinski, M.; Theurillat, J.P.; Nickerson, E.; Auclair, D.; Li, L.; Place, C.; et al. A landscape of driver mutations in melanoma. Cell 2012, 150, 251–263. [Google Scholar] [CrossRef]
- Davies, H.; Bignell, G.R.; Cox, C.; Stephens, P.; Edkins, S.; Clegg, S.; Teague, J.; Woffendin, H.; Garnett, M.J.; Bottomley, W.; et al. Mutations of the BRAF gene in human cancer. Nature 2002, 417, 949–954. [Google Scholar] [CrossRef]
- COSMIC. BRAF. Available online: https://cancer.sanger.ac.uk/cosmic/gene/analysis?ln=BRAF (accessed on 15 August 2023).
- Long, G.V.; Menzies, A.M.; Nagrial, A.M.; Haydu, L.E.; Hamilton, A.L.; Mann, G.J.; Hughes, T.M.; Thompson, J.F.; Scolyer, R.A.; Kefford, R.F. Prognostic and clinicopathologic associations of oncogenic BRAF in metastatic melanoma. J. Clin. Oncol. 2011, 29, 1239–1246. [Google Scholar] [CrossRef]
- COSMIC. NRAS. Available online: https://cancer.sanger.ac.uk/cosmic/gene/analysis?ln=NRAS (accessed on 15 August 2023).
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio Cancer Genomics Portal: An Open Platform for Exploring Multidimensional Cancer Genomics Data. Cancer Discov. 2012, 2, 401. [Google Scholar] [CrossRef] [PubMed]
- cBioPortal. NRAS. Available online: https://www.cbioportal.org/results/cancerTypesSummary?case_set_id=all&gene_list=NRAS&cancer_study_list=5c8a7d55e4b046111fee2296 (accessed on 15 August 2023).
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef] [PubMed]
- Devitt, B.; Liu, W.; Salemi, R.; Wolfe, R.; Kelly, J.; Tzen, C.Y.; Dobrovic, A.; McArthur, G. Clinical outcome and pathological features associated with NRAS mutation in cutaneous melanoma. Pigment Cell Melanoma Res. 2011, 24, 666–672. [Google Scholar] [CrossRef] [PubMed]
- Cook, M.G.; Massi, D.; Blokx, W.A.M.; Van den Oord, J.; Koljenović, S.; De Giorgi, V.; Kissin, E.; Grant, M.; Mandal, A.; Gremel, G.; et al. New insights into naevoid melanomas: A clinicopathological reassessment. Histopathology 2017, 71, 943–950. [Google Scholar] [CrossRef] [PubMed]
- COSMIC. NF1. Available online: https://cancer.sanger.ac.uk/cosmic/gene/analysis?ln=NF1 (accessed on 15 August 2023).
- cBioPortal. NF1. Available online: https://www.cbioportal.org/results/cancerTypesSummary?case_set_id=all&gene_list=NF1&cancer_study_list=5c8a7d55e4b046111fee2296 (accessed on 15 August 2023).
- Philpott, C.; Tovell, H.; Frayling, I.M.; Cooper, D.N.; Upadhyaya, M. The NF1 somatic mutational landscape in sporadic human cancers. Hum. Genom. 2017, 11, 13. [Google Scholar] [CrossRef]
- Wiesner, T.; Kiuru, M.; Scott, S.N.; Arcila, M.; Halpern, A.C.; Hollmann, T.; Berger, M.F.; Busam, K.J. NF1 Mutations Are Common in Desmoplastic Melanoma. Am. J. Surg. Pathol. 2015, 39, 1357–1362. [Google Scholar] [CrossRef]
- Cirenajwis, H.; Lauss, M.; Ekedahl, H.; Törngren, T.; Kvist, A.; Saal, L.H.; Olsson, H.; Staaf, J.; Carneiro, A.; Ingvar, C.; et al. NF1-mutated melanoma tumors harbor distinct clinical and biological characteristics. Mol. Oncol. 2017, 11, 438–451. [Google Scholar] [CrossRef]
- Thielmann, C.M.; Chorti, E.; Matull, J.; Murali, R.; Zaremba, A.; Lodde, G.; Jansen, P.; Richter, L.; Kretz, J.; Möller, I.; et al. NF1-mutated melanomas reveal distinct clinical characteristics depending on tumor origin and respond favorably to immune checkpoint inhibitors. Eur. J. Cancer 2021, 159, 113–124. [Google Scholar] [CrossRef] [PubMed]
- Curtin, J.A.; Busam, K.; Pinkel, D.; Bastian, B.C. Somatic activation of KIT in distinct subtypes of melanoma. J. Clin. Oncol. 2006, 24, 4340–4346. [Google Scholar] [CrossRef] [PubMed]
- Satzger, I.; Schaefer, T.; Kuettler, U.; Broecker, V.; Voelker, B.; Ostertag, H.; Kapp, A.; Gutzmer, R. Analysis of c-KIT expression and KIT gene mutation in human mucosal melanomas. Br. J. Cancer 2008, 99, 2065–2069. [Google Scholar] [CrossRef] [PubMed]
- Gong, H.Z.; Zheng, H.Y.; Li, J. The clinical significance of KIT mutations in melanoma: A meta-analysis. Melanoma Res. 2018, 28, 259–270. [Google Scholar] [CrossRef]
- Pham, D.D.M.; Guhan, S.; Tsao, H. KIT and Melanoma: Biological Insights and Clinical Implications. Yonsei Med. J. 2020, 61, 562–571. [Google Scholar] [CrossRef] [PubMed]
- Siroy, A.E.; Davies, M.A.; Lazar, A.J. The PI3K-AKT Pathway in Melanoma. In Genetics of Melanoma. Cancer Genetics; Torres-Cabala, C., Curry, J., Eds.; Springer: New York, NY, USA, 2016. [Google Scholar] [CrossRef]
- Campbell, P.M.; Der, C.J. Oncogenic Ras and its role in tumor cell invasion and metastasis. Semin. Cancer Biol. 2004, 14, 105–114. [Google Scholar] [CrossRef] [PubMed]
- Paluncic, J.; Kovacevic, Z.; Jansson, P.J.; Kalinowski, D.; Merlot, A.M.; Huang, M.L.; Lok, H.C.; Sahni, S.; Lane, D.J.; Richardson, D.R. Roads to melanoma: Key pathways and emerging players in melanoma progression and oncogenic signaling. Biochim. Biophys. Acta 2016, 1863, 770–784. [Google Scholar] [CrossRef] [PubMed]
- Rascio, F.; Spadaccino, F.; Rocchetti, M.T.; Castellano, G.; Stallone, G.; Netti, G.S.; Ranieri, E. The Pathogenic Role of PI3K/AKT Pathway in Cancer Onset and Drug Resistance: An Updated Review. Cancers 2021, 13, 3949. [Google Scholar] [CrossRef] [PubMed]
- Bucheit, A.B.; Chen, G.; Siroy, A.; Tetzlaff, M.; Broaddus, R.; Milton, D.; Fox, P.; Bassett, R.; Hwu, P.; Gershenwald, J.E.; et al. Complete loss of PTEN protein expression correlates with shorter time to brain metastasis and survival in stage IIIB/C melanoma patients with BRAFV600 mutations. Clin. Cancer Res. 2014, 20, 5527–5536. [Google Scholar] [CrossRef] [PubMed]
- Peng, W.; Chen, J.Q.; Liu, C.; Malu, S.; Creasy, C.; Tetzlaff, M.T.; Xu, C.; McKenzie, J.A.; Zhang, C.; Liang, X.; et al. Loss of PTEN Promotes Resistance to T Cell-Mediated Immunotherapy. Cancer Discov. 2016, 6, 202–216. [Google Scholar] [CrossRef]
- Lucero, O.M.; Dawson, D.W.; Moon, R.T.; Chien, A.J. A re-evaluation of the “oncogenic” nature of Wnt/beta-catenin signaling in melanoma and other cancers. Curr. Oncol. Rep. 2010, 12, 314–318. [Google Scholar] [CrossRef]
- Regad, T. Molecular and cellular pathogenesis of melanoma initiation and progression. Cell. Mol. Life Sci. 2013, 70, 4055–4065. [Google Scholar] [CrossRef] [PubMed]
- Chien, A.J.; Moore, E.C.; Lonsdorf, A.S.; Kulikauskas, R.M.; Rothberg, B.G.; Berger, A.J.; Major, M.B.; Hwang, S.T.; Rimm, D.L.; Moon, R.T. Activated Wnt/beta-catenin signaling in melanoma is associated with decreased proliferation in patient tumors and a murine melanoma model. Proc. Natl. Acad. Sci. USA 2009, 106, 1193–1198. [Google Scholar] [CrossRef]
- Biechele, T.L.; Camp, N.D.; Fass, D.M.; Kulikauskas, R.M.; Robin, N.C.; White, B.D.; Taraska, C.M.; Moore, E.C.; Muster, J.; Karmacharya, R.; et al. Chemical-genetic screen identifies riluzole as an enhancer of Wnt/beta-catenin signaling in melanoma. Chem Biol. 2010, 17, 1177–1182. [Google Scholar] [CrossRef]
- He, S.; Lu, Y.; Liu, X.; Huang, X.; Keller, E.T.; Qian, C.-N.; Zhang, J. Wnt3a: Functions and implications in cancer. Chin. J. Cancer 2015, 34, 50. [Google Scholar] [CrossRef]
- Yeh, I.; Lang, U.E.; Durieux, E.; Tee, M.K.; Jorapur, A.; Shain, A.H.; Haddad, V.; Pissaloux, D.; Chen, X.; Cerroni, L.; et al. Combined activation of MAP kinase pathway and β-catenin signaling cause deep penetrating nevi. Nat. Commun. 2017, 8, 644. [Google Scholar] [CrossRef] [PubMed]
- Luzar, B.; Calonje, E. Deep Penetrating Nevus: A Review. Arch. Pathol. Lab. Med. 2011, 135, 321–326. [Google Scholar] [CrossRef] [PubMed]
- Robson, A.; Morley-Quante, M.; Hempel, H.; McKee, P.H.; Calonje, E. Deep penetrating naevus: Clinicopathological study of 31 cases with further delineation of histological features allowing distinction from other pigmented benign melanocytic lesions and melanoma. Histopathology 2003, 43, 529–537. [Google Scholar] [CrossRef]
- Magro, C.M.; Abraham, R.M.; Guo, R.; Li, S.; Wang, X.; Proper, S.; Crowson, A.N.; Mihm, M. Deep penetrating nevus-like borderline tumors: A unique subset of ambiguous melanocytic tumors with malignant potential and normal cytogenetics. Eur. J. Dermatol. 2014, 24, 594–602. [Google Scholar] [CrossRef] [PubMed]
- Massi, G.; LeBoit, P.E. Histological Diagnosis of Nevi and Melanoma; Springer: Berlin/Heidelberg, Germany, 2014; Melanoma with a plexiform pattern; pp. 581–588. [Google Scholar]
- Isales, M.C.; Khan, A.U.; Zhang, B.; Compres, E.V.; Kim, D.; Tan, T.L.; Beaubier, N.; Gerami, P. Molecular analysis of atypical deep penetrating nevus progressing to melanoma. J. Cutan. Pathol. 2020, 47, 1150–1154. [Google Scholar] [CrossRef] [PubMed]
- Erstine, E.M.; Lazar, A.J. Toward an effective use of β-catenin immunohistochemistry in the evaluation of challenging melanocytic lesions. Virchows Arch. 2019, 474, 535–537. [Google Scholar] [CrossRef]
- Mancarella, D.; Plass, C. Epigenetic Signatures in Cancer: Proper Controls, Current Challenges, and the Potential for Clinical Translation. Genome Med. 2021, 13, 23. [Google Scholar] [CrossRef]
- Fujiwara, S.; Nagai, H.; Jimbo, H.; Jimbo, N.; Tanaka, T.; Inoie, M.; Nishigori, C. Gene Expression and Methylation Analysis in Melanomas and Melanocytes from the Same Patient: Loss of NPM2 Expression Is a Potential Immunohistochemical Marker for Melanoma. Front. Oncol. 2019, 8, 675. [Google Scholar] [CrossRef]
- Aleotti, V.; Catoni, C.; Poggiana, C.; Rosato, A.; Facchinetti, A.; Scaini, M.C. Methylation Markers in Cutaneous Melanoma: Unravelling the Potential Utility of Their Tracking by Liquid Biopsy. Cancers 2021, 13, 6217. [Google Scholar] [CrossRef]
- Sutopo, N.C.; Kim, J.H.; Cho, J.Y. Role of histone methylation in skin cancers: Histone methylation-modifying enzymes as a new class of targets for skin cancer treatment. Biochim. Biophys. Acta. Rev. Cancer 2023, 1878, 188865. [Google Scholar] [CrossRef]
- Kapoor, A.; Goldberg, M.S.; Cumberland, L.K.; Ratnakumar, K.; Segura, M.F.; Emanuel, P.O.; Menendez, S.; Vardabasso, C.; LeRoy, G.; Vidal, C.I.; et al. The histone variant macroH2A suppresses melanoma progression through regulation of CDK8. Nature 2010, 468, 1105–1109. [Google Scholar] [CrossRef] [PubMed]
- Vardabasso, C.; Gaspar-Maia, A.; Hasson, D.; Pünzeler, S.; Valle-Garcia, D.; Straub, T.; Keilhauer, E.C.; Strub, T.; Dong, J.; Panda, T.; et al. Histone variant H2A.Z.2 mediates proliferation and drug sensitivity of malignant melanoma. Mol. Cell 2015, 59, 75–88. [Google Scholar] [CrossRef]
- Giunta, E.F.; Arrichiello, G.; Curvietto, M.; Pappalardo, A.; Bosso, D.; Rosanova, M.; Diana, A.; Giordano, P.; Petrillo, A.; Federico, P.; et al. Epigenetic Regulation in Melanoma: Facts and Hopes. Cells 2021, 10, 2048. [Google Scholar] [CrossRef] [PubMed]
- Palmieri, G.; Colombino, M.; Casula, M.; Manca, A.; Mandalà, M.; Cossu, A.; Italian Melanoma Intergroup (IMI). Molecular Pathways in Melanomagenesis: What We Learned from Next-Generation Sequencing Approaches. Curr. Oncol. Rep. 2018, 20, 86. [Google Scholar] [CrossRef] [PubMed]
- Scatena, C.; Murtas, D.; Tomei, S. Cutaneous Melanoma Classification: The Importance of High-Throughput Genomic Technologies. Front. Oncol. 2021, 11, 635488. [Google Scholar] [CrossRef] [PubMed]
- Diana, A.; Gaido, G.; Murtas, D. Microrna Signature in Human Normal and Tumoral Neural Stem Cells. Int. J. Mol. Sci. 2019, 20, 4123. [Google Scholar] [CrossRef] [PubMed]
- Gajos-Michniewicz, A.; Czyz, M. Role of miRNAs in Melanoma Metastasis. Cancers 2019, 11, 326. [Google Scholar] [CrossRef]
- Lohcharoenkal, W.; Das Mahapatra, K.; Pasquali, L.; Crudden, C.; Kular, L.; Akkaya Ulum, Y.Z.; Zhang, L.; Xu Landén, N.; Girnita, L.; Jagodic, M.; et al. Genome-Wide Screen for MicroRNAs Reveals a Role for miR-203 in Melanoma Metastasis. J. Investig. Dermatol. 2018, 138, 882–892. [Google Scholar] [CrossRef]
- Shah, M.Y.; Ferrajoli, A.; Sood, A.K.; Lopez-Berestein, G.; Calin, G.A. microRNA Therapeutics in Cancer—An Emerging Concept. EBioMedicine 2016, 12, 34–42. [Google Scholar] [CrossRef]
- Varrone, F.; Caputo, E. The miRNAs Role in Melanoma and in Its Resistance to Therapy. Int. J. Mol. Sci. 2020, 21, 878. [Google Scholar] [CrossRef]
- Saleem, A.; Narala, S.; Raghavan, S.S. Immunohistochemistry in melanocytic lesions: Updates with a practical review for pathologists. Semin. Diagn. Pathol. 2022, 39, 239–247. [Google Scholar] [CrossRef] [PubMed]
- Yeh, I. Update on classification of melanocytic tumors and the role of immunohistochemistry and molecular techniques. Semin. Diagn. Pathol. 2022, 39, 248–256. [Google Scholar] [CrossRef]
- Andea, A.A. Molecular testing in melanoma for the surgical pathologist. Pathology 2023, 55, 245–257. [Google Scholar] [CrossRef] [PubMed]
- Quan, V.L.; Panah, E.; Zhang, B.; Shi, K.; Mohan, L.S.; Gerami, P. The role of gene fusions in melanocytic neoplasms. J. Cutan. Pathol. 2019, 46, 878–887. [Google Scholar] [CrossRef] [PubMed]
- Jakob, J.A.; Bassett, R.L., Jr.; Ng, C.S.; Curry, J.L.; Joseph, R.W.; Alvarado, G.C.; Rohlfs, M.L.; Richard, J.; Gershenwald, J.E.; Kim, K.B.; et al. NRAS mutation status is an independent prognostic factor in metastatic melanoma. Cancer 2012, 118, 4014–4023. [Google Scholar] [CrossRef]
- Massi, D.; Simi, L.; Sensi, E.; Baroni, G.; Xue, G.; Scatena, C.; Caldarella, A.; Pinzani, P.; Fontanini, G.; Carobbio, A.; et al. Immunohistochemistry is highly sensitive and specific for the detection of NRASQ61R mutation in melanoma. Mod. Pathol. 2015, 28, 487–497. [Google Scholar] [CrossRef]
- Yeh, I.; de la Fouchardiere, A.; Pissaloux, D.; Mully, T.W.; Garrido, M.C.; Vemula, S.S.; Busam, K.J.; LeBoit, P.E.; McCalmont, T.H.; Bastian, B.C. Clinical, histopathologic, and genomic features of Spitz tumors with ALK fusions. Am. J. Surg. Pathol. 2015, 39, 581–591. [Google Scholar] [CrossRef]
- Raghavan, S.S.; Saleem, A.; Wang, J.Y.; Rieger, K.E.; Brown, R.A.; Novoa, R.A. Diagnostic Utility of LEF1 Immunohistochemistry in Differentiating Deep Penetrating Nevi from Histologic Mimics. Am. J. Surg. Pathol. 2020, 44, 1413–1418. [Google Scholar] [CrossRef]
- Yeh, I. New and evolving concepts of melanocytic nevi and melanocytomas. Mod. Pathol. 2020, 33 (Suppl. S1), 1–14. [Google Scholar] [CrossRef]
- Benton, S.; Zhao, J.; Asadbeigi, S.; Kim, D.; Zhang, B.; Gerami, P. Pigmented Epithelioid Melanocytoma: Morphology and Molecular Drivers. Surg. Pathol. Clin. 2021, 14, 285–292. [Google Scholar] [CrossRef]
- Ricci, C.; Dika, E.; Ambrosi, F.; Lambertini, M.; Veronesi, G.; Barbara, C. Cutaneous Melanomas: A Single Center Experience on the Usage of Immunohistochemistry Applied for the Diagnosis. Int. J. Mol. Sci. 2022, 23, 5911. [Google Scholar] [CrossRef] [PubMed]
- Straume, O.; Sviland, L.; Akslen, L.A. Loss of nuclear p16 protein expression correlates with increased tumor cell proliferation (Ki-67) and poor prognosis in patients with vertical growth phase melanoma. Clin. Cancer Res. 2000, 6, 1845–1853. [Google Scholar] [PubMed]
- Serra, S.; Chetty, R. p16. J. Clin. Pathol. 2018, 71, 853–858. [Google Scholar] [CrossRef] [PubMed]
- Koh, S.S.; Cassarino, D.S. Immunohistochemical Expression of p16 in Melanocytic Lesions: An Updated Review and Meta-analysis. Arch. Pathol. Lab. Med. 2018, 142, 815–828. [Google Scholar] [CrossRef]
- Balaban, G.; Herlyn, M.; Guerry, D.T.; Bartolo, R.C.; Koprowski, H.; Clark, W.H., Jr.; Nowell, P.C. Cytogenetics of human malignant melanoma and premalignant lesions. Cancer Genet. Cytogenet. 1984, 11, 429–439. [Google Scholar] [CrossRef]
- Kallioniemi, A.; Kallioniemi, O.P.; Sudar, D.; Rutovitz, D.; Gray, J.W.; Waldman, F.; Pinkel, D. Comparative genomic hybridization for molecular cytogenetic analysis of solid tumors. Science 1992, 258, 818–821. [Google Scholar] [CrossRef]
- Wiszniewska, J.; Bi, W.; Shaw, C.; Stankiewicz, P.; Kang, S.H.; Pursley, A.N.; Lalani, S.; Hixson, P.; Gambin, T.; Tsai, C.H.; et al. Combined array CGH plus SNP genome analyses in a single assay for optimized clinical testing. Eur. J. Hum. Genet. EJHG 2014, 22, 79–87. [Google Scholar] [CrossRef]
- Bastian, B.C.; LeBoit, P.E.; Hamm, H.; Bröcker, E.B.; Pinkel, D. Chromosomal gains and losses in primary cutaneous melanomas detected by comparative genomic hybridization. Cancer Res. 1998, 58, 2170–2175. [Google Scholar]
- Bastian, B.C.; Olshen, A.B.; LeBoit, P.E.; Pinkel, D. Classifying melanocytic tumors based on DNA copy number changes. Am. J. Pathol. 2003, 163, 1765–1770. [Google Scholar]
- Hosein, A.N.; Song, S.; McCart Reed, A.E.; Jayanthan, J.; Reid, L.E.; Kutasovic, J.R.; Cummings, M.C.; Waddell, N.; Lakhani, S.R.; Chenevix-Trench, G.; et al. Evaluating the repair of DNA derived from formalin-fixed paraffin-embedded tissues prior to genomic profiling by SNP–CGH analysis. Lab. Investig. 2013, 93, 701–710. [Google Scholar] [CrossRef]
- Wang, Y.; Moorhead, M.; Karlin-Neumann, G.; Wang, N.J.; Ireland, J.; Lin, S.; Chen, C.; Heiser, L.M.; Chin, K.; Esserman, L.; et al. Analysis of molecular inversion probe performance for allele copy number determination. Genome Biol. 2007, 8, R246. [Google Scholar] [CrossRef]
- Chandler, W.M.; Rowe, L.R.; Florell, S.R.; Jahromi, M.S.; Schiffman, J.D.; South, S.T. Differentiation of malignant melanoma from benign nevus using a novel genomic microarray with low specimen requirements. Arch. Pathol. Lab. Med. 2012, 136, 947–955. [Google Scholar] [CrossRef] [PubMed]
- Bastian, B.C.; Wesselmann, U.; Pinkel, D.; Leboit, P.E. Molecular cytogenetic analysis of Spitz nevi shows clear differences to melanoma. J. Investig. Dermatol. 1999, 113, 1065–1069. [Google Scholar] [CrossRef] [PubMed]
- Emanuel, P.O.; Andea, A.A.; Vidal, C.I.; Missall, T.A.; Novoa, R.A.; Bohlke, A.K.; Hughes, S.R.; Hurley, M.Y.; Kim, J. Evidence behind the use of molecular tests in melanocytic lesions and practice patterns of these tests by dermatopathologists. J. Cutan. Pathol. 2018, 45, 839–846. [Google Scholar] [CrossRef] [PubMed]
- Jiveskog, S.; Ragnarsson-Olding, B.; Platz, A.; Ringborg, U. N-RAS mutations are common in melanomas from sun-exposed skin of humans but rare in mucosal membranes of unexposed skin. J. Investig. Dermatol. 1998, 111, 757–761. [Google Scholar] [CrossRef]
- Bastian, B.C.; LeBoit, P.E.; Pinkel, D. Mutations and copy number increase of HRAS in Spitz nevi with distinctive histopathological features. Am. J. Pathol. 2000, 157, 967–972. [Google Scholar] [CrossRef]
- Andea, A.A. Molecular testing for melanocytic tumors: A practical update. Histopathology. 2022, 80, 150–165. [Google Scholar] [CrossRef]
- FISH for Melanoma, Multiplex Probe. Available online: https://mlabs.umich.edu/tests/fish-melanoma-multiplex-probe (accessed on 7 January 2023).
- Gerami, P.; Jewell, S.S.; Morrison, L.E.; Blondin, B.; Schulz, J.; Ruffalo, T.; Matushek, P.; Legator, M., IV; Jacobson, K.; Dalton, S.R.; et al. Fluorescence in situ hybridization (FISH) as an ancillary diagnostic tool in the diagnosis of melanoma. Am. J. Surg. Pathol. 2009, 33, 1146–1156. [Google Scholar] [CrossRef]
- Miedema, J.; Andea, A.A. Through the looking glass and what you find there: Making sense of comparative genomic hybridization and fluorescence in situ hybridization for melanoma diagnosis. Mod. Pathol. 2020, 33, 1318–1330. [Google Scholar] [CrossRef]
- Vergier, B.; Prochazkova-Carlotti, M.; de la Fouchardière, A.; Cerroni, L.; Massi, D.; De Giorgi, V.; Bailly, C.; Wesselmann, U.; Karlseladze, A.; Avril, M.F.; et al. Fluorescence in situ hybridization, a diagnostic aid in ambiguous melanocytic tumors: European study of 113 cases. Mod. Pathol. 2011, 24, 613–623. [Google Scholar] [CrossRef]
- Pouryazdanparast, P.; Newman, M.; Mafee, M.; Haghighat, Z.; Guitart, J.; Gerami, P. Distinguishing epithelioid blue nevus from blue nevus-like cutaneous melanoma metastasis using fluorescence in situ hybridization. Am. J. Surg. Pathol. 2009, 33, 1396–1400. [Google Scholar] [CrossRef] [PubMed]
- Ferrara, G.; De Vanna, A.C. Fluorescence In Situ Hybridization for Melanoma Diagnosis: A Review and a Reappraisal. Am. J. Dermatopathol. 2016, 38, 253–269. [Google Scholar] [CrossRef] [PubMed]
- Gerami, P.; Li, G.; Pouryazdanparast, P.; Blondin, B.; Beilfuss, B.; Slenk, C.; Du, J.; Guitart, J.; Jewell, S.; Pestova, K. A highly specific and discriminatory FISH assay for distinguishing between benign and malignant melanocytic neoplasms. Am. J. Surg. Pathol. 2012, 36, 808–817. [Google Scholar] [CrossRef] [PubMed]
- Cho-Vega, J.H.; Cao, T.; Ledon, J.; Moller, M.; Avisar, E.; Elgart, G.; Tan, J.H.; Fan, Y.S.; Grichnik, J.M. Diagnostic application of cyclin D1 fluorescent in situ hybridization for histologically undetermined early lesions of acral melanoma in situ: A case series. Ann. Diagn. Pathol. 2021, 50, 151681. [Google Scholar] [CrossRef]
- Shahbain, H.; Cooper, C.; Gerami, P. Molecular diagnostics for ambiguous melanocytic tumors. Semin. Cutan. Med. Surg. 2012, 31, 274–278. [Google Scholar] [CrossRef]
- Gammon, B.; Beilfuss, B.; Guitart, J.; Gerami, P. Enhanced detection of spitzoid melanomas using fluorescence in situ hybridization with 9p21 as an adjunctive probe. Am. J. Surg. Pathol. 2012, 36, 81–88. [Google Scholar] [CrossRef] [PubMed]
- Curtin, J.A.; Fridlyand, J.; Kageshita, T.; Patel, H.N.; Busam, K.J.; Kutzner, H.; Cho, K.H.; Aiba, S.; Bröcker, E.B.; LeBoit, P.E.; et al. Distinct sets of genetic alterations in melanoma. N. Engl. J. Med. 2005, 353, 2135–2147. [Google Scholar] [PubMed]
- Gerami, P.; Scolyer, R.A.; Xu, X.; Elder, D.E.; Abraham, R.M.; Fullen, D.; Prieto, V.G.; Leboit, P.E.; Barnhill, R.L.; Cooper, C.; et al. Risk assessment for atypical spitzoid melanocytic neo-plasms using FISH to identify chromosomal copy number aberrations. Am. J. Surg. Pathol. 2013, 37, 676–684. [Google Scholar] [CrossRef]
- Cho-Vega, J. A diagnostic algorithm for atypical spitzoid tumors: Guidelines for immunohistochemical and molecular assessment. Mod. Pathol. 2016, 29, 656–670. [Google Scholar] [CrossRef]
- Grossman, D.; Kim, C.C.; Hartman, R.I.; Berry, E.; Nelson, K.C.; Okwundu, N.; Curiel-Lewandrowski, C.; Leachman, S.A.; Swetter, S.M. Prognostic gene expression profiling in melanoma: Necessary steps to incorporate into clinical practice. Melanoma Manag. 2019, 6, MMT32. [Google Scholar] [CrossRef]
- Clarke, L.E.; Warf, M.B.; Flake, D.D., II; Hartman, A.R.; Tahan, S.; Shea, C.R.; Gerami, P.; Messina, J.; Florell, S.R.; Wenstrup, R.J.; et al. Clinical validation of a gene expression signature that differentiates benign nevi from malignant melanoma. J. Cutan. Pathol. 2015, 42, 244–252. [Google Scholar] [CrossRef] [PubMed]
- Yeh, I.; Bastian, B.C. Melanoma pathology: New approaches and classification. Br. J. Dermatol. 2021, 185, 282–293. [Google Scholar] [CrossRef]
- Grossman, D.; Okwundu, N.; Bartlett, E.K.; Marchetti, M.A.; Othus, M.; Coit, D.G.; Hartman, R.I.; Leachman, S.A.; Berry, E.G.; Korde, L.; et al. Prognostic Gene Expression Profiling in Cutaneous Melanoma: Identifying the Knowledge Gaps and Assessing the Clinical Benefit. JAMA Dermatol. 2020, 156, 1004–1011. [Google Scholar] [CrossRef] [PubMed]
- Clarke, L.E.; Mabey, B.; Flake II, D.D.; Meek, S.; Cassarino, D.S.; Duncan, L.M.; High, W.A.; Napekoski, K.M.; Prieto, V.G.; Tetzlaff, M.T.; et al. Clinical validity of a gene expression signature in diagnostically uncertain neoplasms. Pers. Med. 2020, 17, 361–371. [Google Scholar] [CrossRef] [PubMed]
- Greenhaw, B.N.; Covington, K.R.; Kurley, S.J.; Yeniay, Y.; Cao, N.A.; Plasseraud, K.M.; Cook, R.W.; Hsueh, E.C.; Gastman, B.R.; Wei, M.L. Molecular risk prediction in cutaneous melanoma: A meta-analysis of the 31-gene expression profile prognostic test in 1,479 patients. J. Am. Acad. Dermatol. 2020, 83, 745–753. [Google Scholar] [CrossRef] [PubMed]
- Swetter, S.M.; Tsao, H.; Bichakjian, C.K.; Curiel-Lewandrowski, C.; Elder, D.E.; Gershenwald, J.E.; Guild, V.; Grant-Kels, J.M.; Halpern, A.C.; Johnson, T.M.; et al. Guidelines of care for the management of primary cutaneous melanoma. J. Am. Acad. Dermatol. 2019, 80, 208–250. [Google Scholar] [CrossRef] [PubMed]
- Marks, E.; Jarell, A.; Ludzik, J.; Farberg, A.S.; Rabinovitz, H.S.; Phelps, R.G.; Cockerell, C.J.; Witkowski, A. A Physician’s Guide to the Use of Gene Expression Profile Ancillary Diagnostic Testing for Cutaneous Melanocytic Neoplasms. J. Clin. Aesthetic Dermatol. 2023, 16, 12–20. [Google Scholar]
- Heidenreich, B.; Nagore, E.; Rachakonda, P.S.; Garcia-Casado, Z.; Requena, C.; Traves, V.; Becker, J.; Soufir, N.; Hemminki, K.; Kumar, R. Telomerase reverse transcriptase promoter mutations in primary cutaneous melanoma. Nat. Commun. 2014, 5, 3401. [Google Scholar] [CrossRef]
- Osella-Abate, S.; Bertero, L.; Senetta, R.; Mariani, S.; Lisa, F.; Coppola, V.; Metovic, J.; Pasini, B.; Puig, S.S.; Fierro, M.T.; et al. TERT Promoter Mutations are Associated with Visceral Spreading in Melanoma of the Trunk. Cancers 2019, 11, 452. [Google Scholar] [CrossRef]
- Macerola, E.; Loggini, B.; Giannini, R.; Garavello, G.; Giordano, M.; Proietti, A.; Niccoli, C.; Basolo, F.; Fontanini, G. Coexistence of TERT promoter and BRAF mutations in cutaneous melanoma is associated with more clinicopathological features of aggressiveness. Virchows Arch. 2015, 467, 177–184. [Google Scholar] [CrossRef]
- Neumann, E.K.; Djambazova, K.V.; Caprioli, R.M.; Spraggins, J.M. Multimodal Imaging Mass Spectrometry: Next Generation Molecular Mapping in Biology and Medicine. J. Am. Soc. Mass Spectrom. 2020, 31, 2401–2415. [Google Scholar] [CrossRef] [PubMed]
- Ryan, D.J.; Spraggins, J.M.; Caprioli, R.M. Protein identification strategies in MALDI imaging mass spectrometry: A brief review. Curr. Opin. Chem. Biol. 2019, 48, 64–72. [Google Scholar] [CrossRef]
- Casadonte, R.; Kriegsmann, M.; Kriegsmann, K.; Hauk, I.; Meliß, R.R.; Müller, C.S.L.; Kriegsmann, J. Imaging Mass Spectrometry-Based Proteomic Analysis to Differentiate Melanocytic Nevi and Malignant Melanoma. Cancers 2021, 13, 3197. [Google Scholar] [CrossRef] [PubMed]
- Lazova, R.; Seeley, E.H.; Keenan, M.; Gueorguieva, R.; Caprioli, R.M. Imaging mass spectrometry—A new and promising method to differentiate Spitz nevi from Spitzoid malignant melanomas. Am. J. Dermatopathol. 2012, 34, 82–90. [Google Scholar] [CrossRef] [PubMed]
- Hardesty, W.M.; Caprioli, R.M. In situ molecular imaging of proteins in tissues using mass spectrometry. Anal. Bioanal. Chem. 2008, 391, 899–903. [Google Scholar] [CrossRef] [PubMed]
- Hardesty, W.M.; Kelley, M.C.; Mi, D.; Low, R.L.; Caprioli, R.M. Protein signatures for survival and recurrence in metastatic melanoma. J. Proteom. 2011, 74, 1002–1014. [Google Scholar] [CrossRef] [PubMed]
- Casadonte, R.; Kriegsmann, M.; Kriegsmann, K.; Streit, H.; Meliß, R.R.; Müller, C.S.L.; Kriegsmann, J. Imaging Mass Spectrometry for the Classification of Melanoma Based on BRAF/NRAS Mutational Status. Int. J. Mol. Sci. 2023, 24, 5110. [Google Scholar] [CrossRef]
- Harvard Cancer Center. Available online: https://www.dfhcc.harvard.edu/research/core-facilities/cancer-proteomics/pricing (accessed on 3 January 2024).
- Cree, I.A.; Bastian, B.C.; Barnhill, R.L.; Elder, D.E.; Lazar, A.J.; Scolyer, R.A.; de la Fouchardière, A.; Elder, D.E.; Gerami, P.; Massi, D.; et al. Melanocytic neoplasms: Genomic landscape of melanoma. In WHO Classification of Tumours Editorial Board. Skin Tumours [Internet; Beta Version Ahead of Print], 5th ed.; WHO Classification of Tumours Series; International Agency for Research on Cancer: Lyon, France, 2023; Volume 12. Available online: https://tumourclassification.iarc.who.int/chapters/64 (accessed on 25 October 2023).
- Lee, E.Y.; Williamson, R.; Watt, P.; Hughes, M.C.; Green, A.C.; Whiteman, D.C. Sun exposure and host phenotype as predictors of cutaneous melanoma associated with neval remnants or dermal elastosis. Int. J. Cancer. 2006, 119, 636–642. [Google Scholar] [CrossRef]
- Connolly, K.L.; Nehal, K.S.; Busam, K.J. Lentigo Maligna and Lentigo Maligna Melanoma: Contemporary Issues in Diagnosis and Management. Melanoma Manag. 2015, 2, 171–178. [Google Scholar] [CrossRef]
- Loras, A.; Gil-Barrachina, M.; Marqués-Torrejón, M.Á.; Perez-Pastor, G.; Martinez-Cadenas, C. UV-Induced Somatic Mutations Driving Clonal Evolution in Healthy Skin, Nevus, and Cutaneous Melanoma. Life 2022, 12, 1339. [Google Scholar] [CrossRef]
- Cherepakhin, O.S.; Argenyi, Z.B.; Moshiri, A.S. Genomic and Transcriptomic Underpinnings of Melanoma Genesis, Progression, and Metastasis. Cancers 2021, 14, 123. [Google Scholar] [CrossRef] [PubMed]
- Lázár, V.; Ecsedi, S.; Szöllosi, A.G.; Tóth, R.; Vízkeleti, L.; Rákosy, Z.; Bégány, A.; Adány, R.; Balázs, M. Characterization of candidate gene copy number alterations in the 11q13 region along with BRAF and NRAS mutations in human melanoma. Mod. Pathol. 2009, 22, 1367–1378. [Google Scholar] [CrossRef] [PubMed]
- Sanna, A.; Harbst, K.; Johansson, I.; Christensen, G.; Lauss, M.; Mitra, S.; Rosengren, F.; Häkkinen, J.; Vallon-Christersson, J.; Olsson, H.; et al. Tumor Genetic Heterogeneity Analysis of Chronic Sun-Damaged Melanoma. Pigment Cell Melanoma Res. 2020, 33, 480–489. [Google Scholar] [CrossRef] [PubMed]
- Bobos, M. Histopathologic Classification and Prognostic Factors of Melanoma: A 2021 Update. Ital. J. Dermatol. Venerol. 2021, 156, 300–321. [Google Scholar] [CrossRef]
- Tímár, J.; Vizkeleti, L.; Doma, V.; Barbai, T.; Rásó, E. Genetic Progression of Malignant Melanoma. Cancer Metastasis Rev. 2016, 35, 93–107. [Google Scholar] [CrossRef] [PubMed]
- Mar, V.J.; Wong, S.Q.; Li, J.; Scolyer, R.A.; McLean, C.; Papenfuss, A.T.; Tothill, R.W.; Kakavand, H.; Mann, G.J.; Thompson, J.F.; et al. BRAF/NRAS wild-type melanomas have a high mutation load correlating with histological and molecular signatures of UV damage. Clin. Cancer Res. 2013, 19, 4589–4598. [Google Scholar] [CrossRef] [PubMed]
- Goodman, A.M.; Kato, S.; Bazhenova, L.; Patel, S.P.; Frampton, G.M.; Miller, V.; Stephens, P.J.; Daniels, G.A.; Kurzrock, R. Tumor mutational burden as an independent predictor of response to immunotherapy in diverse cancers. Mol. Cancer Ther. 2017, 16, 2598–2608. [Google Scholar] [CrossRef]
- Feng, Z.; Wu, X.; Chen, V.; Velie, E.; Zhang, Z. Incidence and survival of desmoplastic melanoma in the United States, 1992-2007. J. Cutan. Pathol. 2011, 38, 616–624. [Google Scholar] [CrossRef]
- Shain, A.H.; Garrido, M.; Botton, T.; Talevich, E.; Yeh, I.; Sanborn, J.Z.; Chung, J.; Wang, N.J.; Kakavand, H.; Mann, G.J.; et al. Exome sequencing of desmoplastic melanoma identifies recurrent NFKBIE promoter mutations and diverse activating mutations in the MAPK pathway. Nat. Genet. 2015, 47, 1194–1199. [Google Scholar] [CrossRef]
- Elder, D.E.; Bastian, B.C.; Cree, I.A.; Massi, D.; Scolyer, R.A. The 2018 World Health Organization Classification of Cutaneous, Mucosal, and Uveal Melanoma: Detailed Analysis of 9 Distinct Subtypes Defined by Their Evolutionary Pathway. Arch. Pathol. Lab. Med. 2020, 144, 500–522. [Google Scholar] [CrossRef]
- Ostrowski, S.M.; Fisher, D.E. Biology of Melanoma. Hematol. Oncol. Clin. N. Am. 2021, 35, 29–56. [Google Scholar] [CrossRef] [PubMed]
- Krauthammer, M.; Kong, Y.; Bacchiocchi, A.; Evans, P.; Pornputtapong, N.; Wu, C.; McCusker, J.P.; Ma, S.; Cheng, E.; Straub, R.; et al. Exome sequencing identifies recurrent mutations in NF1 and RASopathy genes in sun-exposed melanomas. Nat. Genet. 2015, 47, 996–1002. [Google Scholar] [CrossRef] [PubMed]
- Paniago-Pereira, C.; Maize, J.C.; Ackerman, A.B. Nevus of large spindle and/or epithelioid cells (Spitz’s nevus). Arch. Dermatol. 1978, 114, 1811–1823. [Google Scholar] [CrossRef] [PubMed]
- Newman, S.; Fan, L.; Pribnow, A.; Silkov, A.; Rice, S.V.; Lee, S.; Shao, Y.; Shaner, B.; Mulder, H.; Nakitandwe, J.; et al. Clinical genome sequencing uncovers potentially targetable truncations and fusions of MAP3K8 in spitzoid and other melanomas. Nat. Med. 2019, 25, 597–602. [Google Scholar] [CrossRef] [PubMed]
- Wiesner, T.; He, J.; Yelensky, R.; Esteve-Puig, R.; Botton, T.; Yeh, I.; Lipson, D.; Otto, G.; Brennan, K.; Murali, R.; et al. Kinase fusions are frequent in Spitz tumours and spitzoid melanomas. Nat. Commun. 2014, 5, 3116. [Google Scholar] [CrossRef]
- Yeh, I.; Botton, T.; Talevich, E.; Shain, A.H.; Sparatta, A.J.; de la Fouchardiere, A.; Mully, T.W.; North, J.P.; Garrido, M.C.; Gagnon, A.; et al. Activating MET kinase rearrangements in melanoma and Spitz tumours. Nat. Commun. 2015, 6, 7174. [Google Scholar] [CrossRef]
- Newman, S.; Pappo, A.; Raimondi, S.; Zhang, J.; Barnhill, R.; Bahrami, A. Pathologic Characteristics of Spitz Melanoma with MAP3K8 Fusion or Truncation in a Pediatric Cohort. Am. J. Surg. Pathol. 2019, 43, 1631–1637. [Google Scholar] [CrossRef]
- Amin, S.M.; Haugh, A.M.; Lee, C.Y.; Zhang, B.; Bubley, J.A.; Merkel, E.A.; Verzì, A.E.; Gerami, P. A comparison of morphologic and molecular features of BRAF, ALK, and NTRK1 fusion Spitzoid neoplasms. Am. J. Surg. Pathol. 2017, 41, 491–498. [Google Scholar]
- Busam, K.J.; Kutzner, H.; Cerroni, L.; Wiesner, T. Clinical and pathologic findings of Spitz nevi and atypical Spitz tumors with ALK fusions. Am. J. Surg. Pathol. 2014, 38, 925–933. [Google Scholar] [CrossRef]
- Yeh, I.; Busam, K.J.; McCalmont, T.H.; LeBoit, P.E.; Pissaloux, D.; Alberti, L.; de la Fouchardière, A.; Bastian, B.C. Filigree-like rete ridges, lobulated nests, rosette-like structures, and exaggerated maturation characterize Spitz tumors with NTRK1 fusion. Am. J. Surg. Pathol. 2019, 43, 737–746. [Google Scholar] [CrossRef]
- Quan, V.L.; Zhang, B.; Mohan, L.S.; Shi, K.; Isales, M.C.; Panah, E.; Taxter, T.J.; Beaubier, N.; White, K.; Gerami, P. Activating structural alterations in MAPK genes are distinct genetic drivers in a unique subgroup of Spitzoid neoplasms. Am. J. Surg. Pathol. 2019, 43, 538–548. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Barnhill, R.L.; Dummer, R.; Dalton, J.; Wu, J.; Pappo, A.; Bahrami, A. TERT Promoter Mutations Are Predictive of Aggressive Clinical Behavior in Patients with Spitzoid Melanocytic Neoplasms. Sci. Rep. 2015, 5, 11200. [Google Scholar] [CrossRef] [PubMed]
- Hillen, L.M.; Van den Oord, J.; Geybels, M.S.; Becker, J.C.; Zur Hausen, A.; Winnepenninckx, V. Genomic Landscape of Spitzoid Neoplasms Impacting Patient Management. Front. Med. 2018, 5, 344. [Google Scholar] [CrossRef] [PubMed]
- Al Dhaybi, R.; Agoumi, M.; Gagné, I.; McCuaig, C.; Powell, J.; Kokta, V. P16 Expression: A Marker of Differentiation Between Childhood Malignant Melanomas and Spitz Nevi. J. Am. Acad. Dermatol. 2011, 65, 357–363. [Google Scholar] [CrossRef] [PubMed]
- Hilliard, N.J.; Krahl, D.; Sellheyer, K. P16 Expression Differentiates Between Desmoplastic Spitz Nevus and Desmoplastic Melanoma. J. Cutan. Pathol. 2009, 367, 753–759. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Benton, S.; Zhang, B.; Olivares, S.; Asadbeigi, S.; Busam, K.; Gerami, P. Benign and Intermediate-grade Melanocytic Tumors with BRAF Mutations and Spitzoid Morphology: A Subset of Melanocytic Neoplasms Distinct From Melanoma. Am. J. Surg. Pathol. 2022, 46, 476–485. [Google Scholar] [CrossRef]
- Whiteman, D.C.; Pavan, W.J.; Bastian, B.C. The melanomas: A synthesis of epidemiological, clinical, histopathological, genetic, and biological aspects, supporting distinct subtypes, causal pathways, and cells of origin. Pigment. Cell Melanoma Res. 2011, 24, 879–897. [Google Scholar] [CrossRef]
- Durbec, F.; Martin, L.; Derancourt, C.; Grange, F. Melanoma of the hand and foot: Epidemiological, prognostic and genetic features. A systematic review. Br. J. Dermatol. 2011, 166, 727–739. [Google Scholar] [CrossRef]
- Bastian, B.C. The Molecular Pathology of Melanoma: An Integrated Taxonomy of Melanocytic Neoplasia. Annu. Rev. Pathol. Mech. Dis. 2014, 9, 239–271. [Google Scholar] [CrossRef]
- Chang, J.W. Acral melanoma: A unique disease in Asia. JAMA Dermatol. 2013, 149, 1272–1273. [Google Scholar] [CrossRef]
- Lee, J.H.; Choi, Y.D.; Hwang, J.H.; Shin, M.H.; Yun, S.J. Frequency of Trauma, Physical Stress, and Occupation in Acral Melanoma: Analysis of 313 Acral Melanoma Patients in Korea. Ann. Dermatol. 2021, 33, 228–236. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, N.; Aoto, T.; Uhara, H.; Yamazaki, S.; Akutsu, H.; Umezawa, A.; Nakauchi, H.; Miyachi, Y.; Saida, T.; Nishimura, E.K. A melanocyte—Melanoma precursor niche in sweat glands of volar skin. Pigment Cell Melanoma Res. 2014, 27, 1039–1050. [Google Scholar] [CrossRef] [PubMed]
- Basurto-Lozada, P.; Molina-Aguilar, C.; Castaneda-Garcia, C.; Vázquez-Cruz, M.E.; Garcia-Salinas, O.I.; Álvarez-Cano, A.; Martínez-Said, H.; Roldán-Marín, R.; Adams, D.J.; Possik, P.A.; et al. Acral lentiginous melanoma: Basic facts, biological characteristics and research perspectives of an understudied disease. Pigment Cell Melanoma Res. 2021, 34, 59–71. [Google Scholar] [CrossRef]
- COSMIC. Available online: https://cancer.sanger.ac.uk/cosmic/browse/tissue?wgs=off&sn=skin&ss=NS&hn=malignant_melanoma&sh=acral_lentiginous&in=t&src=tissue&all_data=n (accessed on 18 July 2023).
- Liu, J.; Yu, W.; Gao, F.; Qi, S.; Du, J.; Ma, X.; Zhang, Y.; Zheng, J.; Su, J. CCND1 copy number increase and cyclin D1 expression in acral melanoma: A comparative study of fluorescence in situ hybridization and immunohistochemistry in a Chinese cohort. Diagn. Pathol. 2021, 16, 60. [Google Scholar] [CrossRef] [PubMed]
- Chang, A.E.; Karnell, L.H.; Menck, H.R. The National Cancer Data Base report on cutaneous and noncutaneous melanoma: A summary of 84,836 cases from the past decade. The American College of Surgeons Commission on Cancer and the American Cancer Society. Cancer 1998, 83, 1664–1678. [Google Scholar] [CrossRef]
- Bishop, K.D.; Olszewski, A.J. Epidemiology and survival outcomes of ocular and mucosal melanomas: A population-based analysis. Int. J. Cancer. 2014, 134, 2961–2971. [Google Scholar] [CrossRef] [PubMed]
- Williams, M.D. Update from the 4th Edition of the World Health Organization Classification of Head and Neck Tumours: Mucosal Melanomas. Head Neck Pathol. 2017, 11, 110–117. [Google Scholar] [CrossRef] [PubMed]
- Newell, F.; Kong, Y.; Wilmott, J.S.; Johansson, P.A.; Ferguson, P.M.; Cui, C.; Li, Z.; Kazakoff, S.H.; Burke, H.; Dodds, T.J.; et al. Whole-genome landscape of mucosal melanoma reveals diverse drivers and therapeutic targets. Nat. Commun. 2019, 10, 3163. [Google Scholar] [CrossRef]
- Ciccarese, G.; Drago, F.; Broccolo, F.; Pastorino, A.; Pizzatti, L.; Atzori, L.; Pilloni, L.; Santinelli, D.; Urbani, A.; Parodi, A.; et al. Oncoviruses and melanomas: A retrospective study and literature review. J. Med. Virol. 2023, 95, e27924. [Google Scholar] [CrossRef]
- Furney, S.J.; Turajlic, S.; Stamp, G.; Nohadani, M.; Carlisle, A.; Thomas, J.M.; Hayes, A.; Strauss, D.; Gore, M.; van den Oord, J.; et al. Genome sequencing of mucosal melanomas reveals that they are driven by distinct mechanisms from cutaneous melanoma. J. Pathol. 2013, 230, 261–269. [Google Scholar] [CrossRef]
- Ablain, J.; Xu, M.; Rothschild, H.; Jordan, R.C.; Mito, J.K.; Daniels, B.H.; Bell, C.F.; Joseph, N.M.; Wu, H.; Bastian, B.C.; et al. Human tumor genomics and zebrafish modeling identify SPRED1 loss as a driver of mucosal melanoma. Science 2018, 362, 1055–1060. [Google Scholar] [CrossRef] [PubMed]
- Kinsler, V.A.; O’Hare, P.; Bulstrode, N.; Calonje, J.E.; Chong, W.K.; Hargrave, D.; Jacques, T.; Lomas, D.; Sebire, N.J.; Slater, O. Melanoma in congenital melanocytic naevi. Br. J. Dermatol. 2017, 176, 1131–1143. [Google Scholar] [CrossRef] [PubMed]
- Navarro-Fernandez, I.N.; Mahabal, G.D. Congenital Nevus. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2023. [Google Scholar]
- Bauer, J.; Curtin, J.A.; Pinkel, D.; Bastian, B.C. Congenital melanocytic nevi frequently harbor NRAS mutations but no BRAF mutations. J. Investig. Dermatol. 2007, 127, 179–182. [Google Scholar] [PubMed]
- Ichii-Nakato, N.; Takata, M.; Takayanagi, S.; Takashima, S.; Lin, J.; Murata, H.; Fujimoto, A.; Hatta, N.; Saida, T. High frequency of BRAFV600E mutation in acquired nevi and small congenital nevi, but low frequency of mutation in medium-sized congenital nevi. J. Investig. Dermatol. 2006, 126, 2111–2118. [Google Scholar] [CrossRef]
- Lu, C.; Zhang, J.; Nagahawatte, P.; Easton, J.; Lee, S.; Liu, Z.; Ding, L.; Wyczalkowski, M.A.; Valentine, M.; Navid, F.; et al. The genomic landscape of childhood and adolescent melanoma. J. Investig. Dermatol. 2015, 135, 816–823. [Google Scholar] [CrossRef] [PubMed]
- Akbani, R.; Akdemir, K.C.; Aksoy, B.A.; Albert, M.; Ally, A.; Amin, S.B.; Arachchi, H.; Arora, A.; Auman, J.T.; Ayala, B.; et al. Cancer Genome Atlas Network. Genomic Classification of Cutaneous Melanoma. Cell 2015, 161, 1681–1696. [Google Scholar] [CrossRef]
- Baltres, A.; Salhi, A.; Houlier, A.; Pissaloux, D.; Tirode, F.; Haddad, V.; Karanian, M.; Ysmail-Dahlouk, S.; Boukendakdji, F.; Dahlouk, D.; et al. Malignant melanoma with areas of rhabdomyosarcomatous differentiation arising in a giant congenital nevus with RAF1 gene fusion. Pigment Cell Melanoma Res. 2019, 32, 708–713. [Google Scholar] [CrossRef]
- Harou, O.; Tondeur, G.; Descotes, F.; Balme, B.; Depaepe, L.; Bringuier, P.-P.; Caramel, J.; Thomas, L.; Dalle, S.; Lopez, J. The dynamic molecular landscape of malignant melanomas arising from congenital or common nevi. Integr. Mol. Med. 2019, 6, 1–4. [Google Scholar] [CrossRef]
- Adameyko, I.; Lallemend, F.; Aquino, J.B.; Pereira, J.A.; Topilko, P.; Müller, T.; Fritz, N.; Beljajeva, A.; Mochii, M.; Liste, I.; et al. Schwann cell precursors from nerve innervation are a cellular origin of melanocytes in skin. Cell 2009, 139, 366–379. [Google Scholar] [CrossRef]
- Van Raamsdonk, C.D.; Bezrookove, V.; Green, G.; Bauer, J.; Gaugler, L.; O’Brien, J.M.; Simpson, E.M.; Barsh, G.S.; Bastian, B.C. Frequent somatic mutations of GNAQ in uveal melanoma and blue naevi. Nature 2009, 457, 599–602. [Google Scholar] [CrossRef]
- Möller, I.; Murali, R.; Müller, H.; Wiesner, T.; Jackett, L.A.; Scholz, S.L.; Cosgarea, I.; van de Nes, J.A.; Sucker, A.; Hillen, U.; et al. Activating cysteinyl leukotriene receptor 2 (CYSLTR2) mutations in blue nevi. Mod. Pathol. 2017, 30, 350–356. [Google Scholar] [CrossRef] [PubMed]
- Goto, K.; Pissaloux, D.; Paindavoine, S.; Tirode, F.; de la Fouchardière, A. CYSLTR2-mutant Cutaneous Melanocytic Neoplasms Frequently Simulate “Pigmented Epithelioid Melanocytoma”, Expanding the Morphologic Spectrum of Blue Tumors: A Clinicopathologic Study of 7 Cases. Am. J. Surg. Pathol. 2019, 43, 1368–1376. [Google Scholar] [CrossRef] [PubMed]
- Costa, S.; Byrne, M.; Pissaloux, D.; Haddad, V.; Paindavoine, S.; Thomas, L.; Aubin, F.; Lesimple, T.; Grange, F.; Bonniaud, B.; et al. Melanomas Associated with Blue Nevi or Mimicking Cellular Blue Nevi: Clinical, Pathologic, and Molecular Study of 11 Cases Displaying a High Frequency of GNA11 Mutations, BAP1 Expression Loss, and a Predilection for the Scalp. Am. J. Surg. Pathol. 2016, 40, 368–377. [Google Scholar] [CrossRef] [PubMed]
- Griewank, K.G.; Müller, H.; Jackett, L.A.; Emberger, M.; Möller, I.; van de Nes, J.A.; Zimmer, L.; Livingstone, E.; Wiesner, T.; Scholz, S.L.; et al. SF3B1 and BAP1 mutations in blue nevus-like melanoma. Mod. Pathol. 2017, 30, 928–939. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| IHC Stain | Molecular Alteration Detected | Types of Melanocytic Tumors in Which It Can Be Found | Role in Diagnosis |
|---|---|---|---|
| BRAF V600E (VE1) | BRAF V600E activating point mutation |
| Differentiating between: superficial spreading melanoma with spitzoid morphology (+) vs. low-risk Spitz lesions (−) |
| NRAS Q61R | NRAS Q61R activating point mutation |
| Differentiating between: superficial spreading melanoma with spitzoid morphology (+) vs. low-risk Spitz lesions (−) |
| ALK | ALK translocation | Spitz tumor | Diagnosis of ALK-rearranged Spitz tumors (+) |
| ROS1 | ROS1 translocation | Spitz tumor | Diagnosis of ROS1-rearranged Spitz tumors (+) |
| Pan-TRK | NTRK1 or NTRK3 translocation | Spitz tumor | Diagnosis of NTRK-rearranged Spitz tumors (+) |
| β-catenin/LEF1 | CTNNB1 activating mutation |
| Diagnosis of deep penetrating nevus (nuclear (+)) |
| BAP1 | Loss of function mutation and loss of heterozygosity in BAP1 | BAP1-inactivated melanocytic tumor | Differentiating between: BAP1 inactivated melanocytic tumor (−) vs. Spitz tumor (+) |
| PRKR1A1 | Loss of function mutation and loss of heterozygosity in PRKR1A1 | Pigmented epithelioid melanocytoma | Diagnosis of pigmented epithelioid melanocytoma (−) |
| p16 | CDKN2A biallelic inactivation | Melanoma |
|
| Molecular Platform | Application Scenarios | Pros | Cons |
|---|---|---|---|
| CGH/SNP | Spitz with tetraploidy, superior to FISH | Covers entire genomeHigher sensitivity than FISH | Melanoma infiltrated by other cells can confound the test |
| FISH |
|
|
|
| Gene expression profiling | Diagnosis and prognosis of melanocytic tumors | Minimal cells required for diagnosis: tape stripping from surface of pigmented lesion is sufficient |
|
| Mutational analysis: TERT-p and BRAF |
| TERT-p mutation has a high specificity for differentiating melanomas from nevi | TERT-p mutation has a low sensitivity for diagnosing melanoma |
| MALDI-IMS |
|
| Not readily available in most laboratories |
| Pathways | Low CSD (1) | High CSD (2) | DM (3) | Spitz (4) | Acral (5) | Mucosal (6) | Melanoma Arising from CN (7) | MBN (8) |
|---|---|---|---|---|---|---|---|---|
| Initial molecular alteration(s) | BRAF V600E NRAS PTEN | NRAS BRAF(non-V600) KIT RASA2 | NF1 PIK3CA NRAS | HRAS Kinase fusion: ALK, NTRK1, ROS1, BRAF, MET | CCND1 KIT | CCND1 KIT BRAF | CNAs NRAS: giant CN BRAF: medium-to-small CN | GNAQ GNA11 CYSLTR1 |
| Additional molecular alterations | CDKN2A TP53 PTEN TERT-p | TP53 NF1 ARID2 CCND1 CDKN2A PTEN TERT-p | CDKN2A BRAF fusion MAP3K8 fusion TERT-p | TERT-p, CDKN2A | NRAS NF1 CDKN2A SPRED1 SF3B1 TERT-p | KIT PTEN CDKN2A TP53 TERT-p | BAP1 SF3B1 | |
| Useful IHCs | BRAFV 600E (VE1) NRASQ61R p16 | NRAS Q61R c-kit | NRAS Q61R | ROS1 ALK pan-TRK p16 | p16 | p16 | NRASQ61R p16 | BAP1 |
| Useful molecular tests | Mutation analysis for BRAF V600E and TERT-p | CGH FISH Mutation analysis for TERT-p | Usually not needed | CGH FISH | FISH for CCND1 amplification | FISH | FISH Mutation analysis for TERT-p | FISH |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, C.; Lau, T.W.-S.; Smoller, B.R. Diagnosing Cutaneous Melanocytic Tumors in the Molecular Era: Updates and Review of Literature. Dermatopathology 2024, 11, 26-51. https://doi.org/10.3390/dermatopathology11010005
Huang C, Lau TW-S, Smoller BR. Diagnosing Cutaneous Melanocytic Tumors in the Molecular Era: Updates and Review of Literature. Dermatopathology. 2024; 11(1):26-51. https://doi.org/10.3390/dermatopathology11010005
Chicago/Turabian StyleHuang, Chelsea, Tiffany Wing-See Lau, and Bruce R. Smoller. 2024. "Diagnosing Cutaneous Melanocytic Tumors in the Molecular Era: Updates and Review of Literature" Dermatopathology 11, no. 1: 26-51. https://doi.org/10.3390/dermatopathology11010005