Development and Permeability Testing of Self-Emulsifying Atorvastatin Calcium Pellets and Tablets of Compressed Pellets


Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Methods
2.2.1. Screening Tests
2.2.2. HPLC Analysis
2.2.3. Preparation of Nanoemulsion-Ultrasonication
2.2.4. Characterization of Emulsion
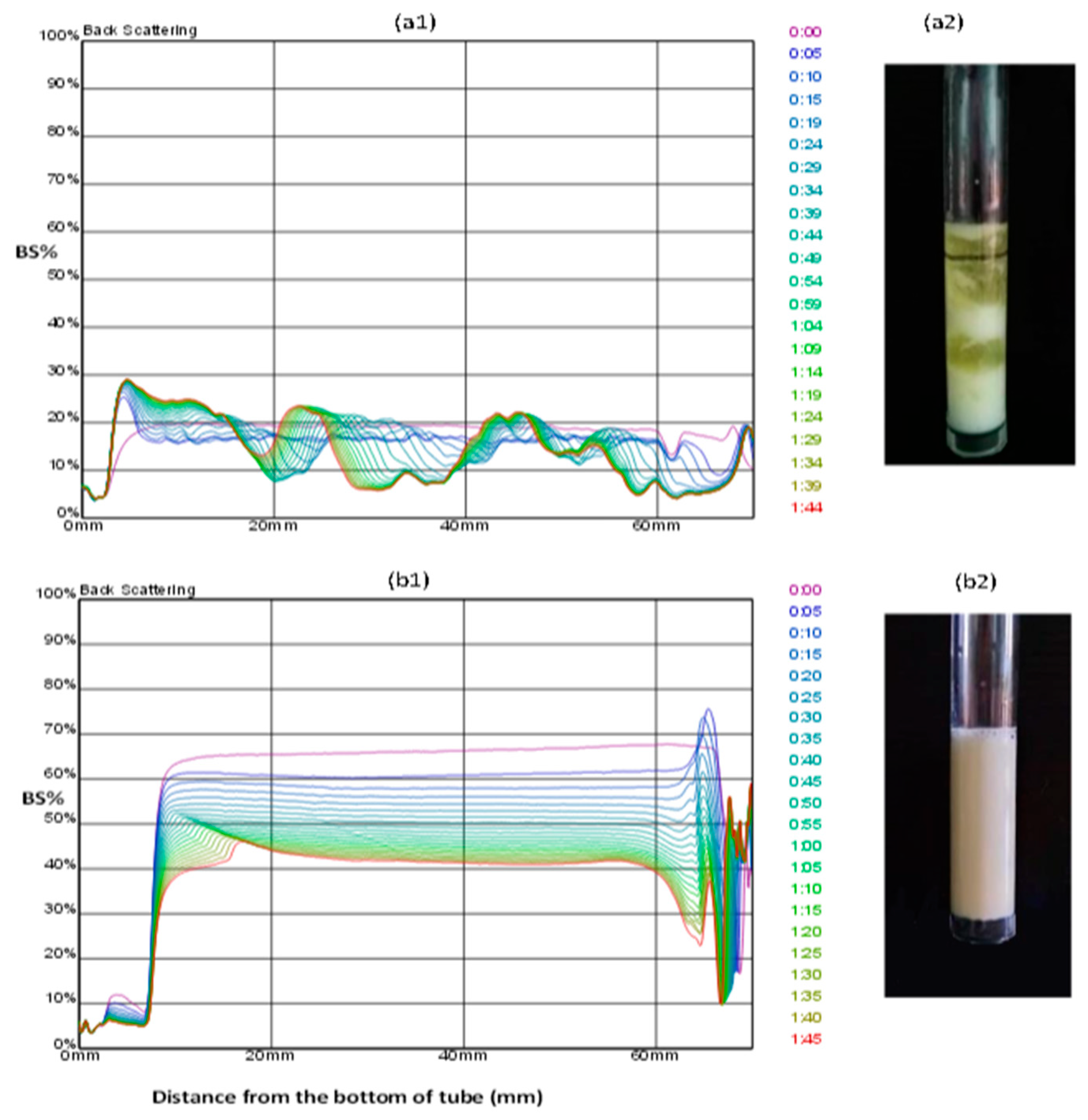
2.2.5. Emulsion stability
2.2.6. Preparation of Self Emulsifying Pellets (SEPs)
2.2.7. Chemical Stability
2.2.8. Characterization of Self Emulsifying Pellets
Pycnometric and tap densities
Particle Size and Shape Analysis
2.2.9. Reconstitution of Emulsions from Pellets
2.2.10. Preparation of Self-Emulsifying Tablets (SETs)
2.2.11. Evaluation of SETs
2.2.12. In Vitro Release
2.2.13. Permeability experiments-Caco-2 Cells
2.2.14. Cytotoxicity Test
3. Results and discussion
3.1. Screening Studies
3.2. Preparation
3.3. Characterization of Microemulsions
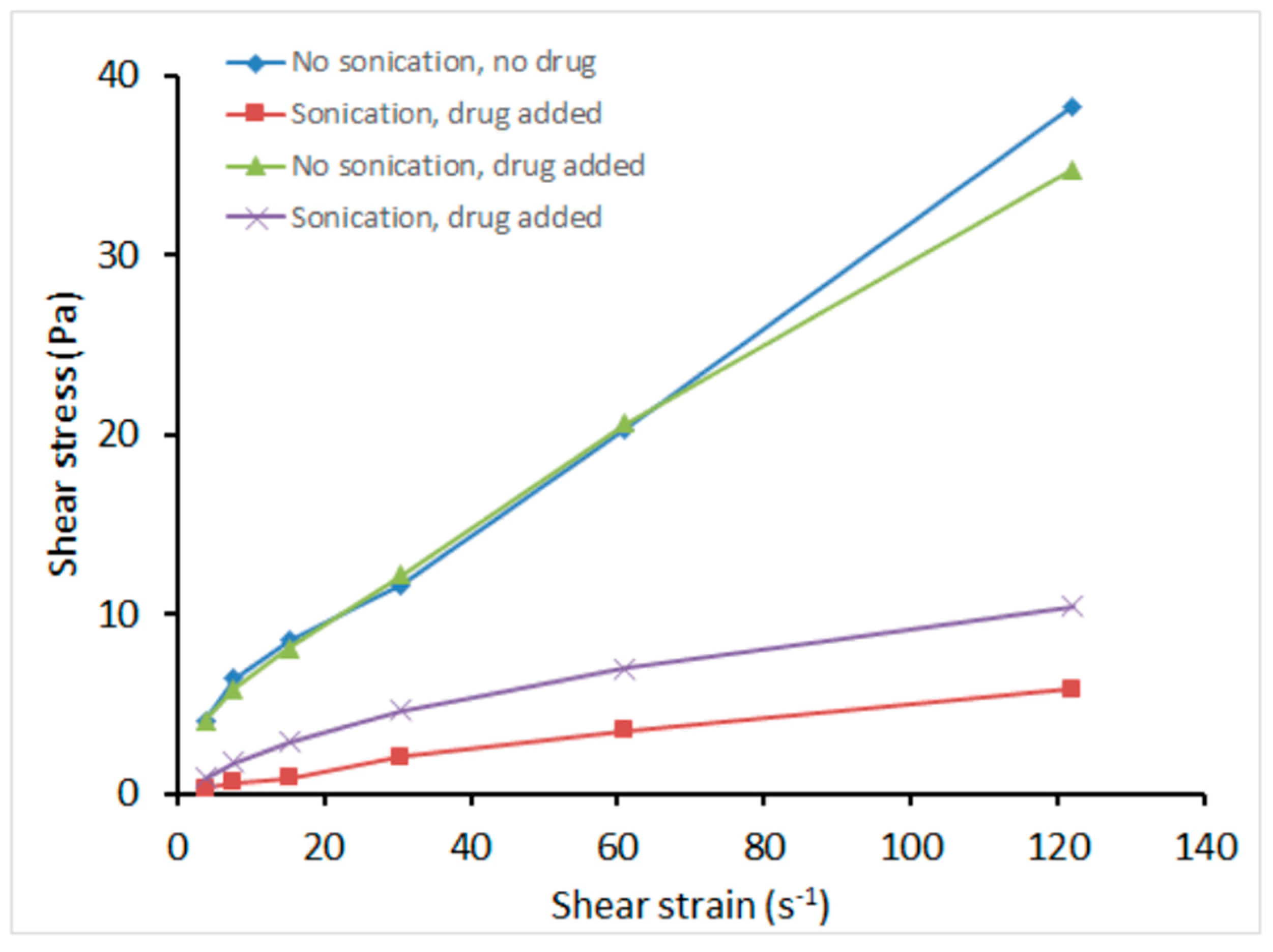
3.3.1. Rheology
3.3.2. Droplet Size and Zeta Potential
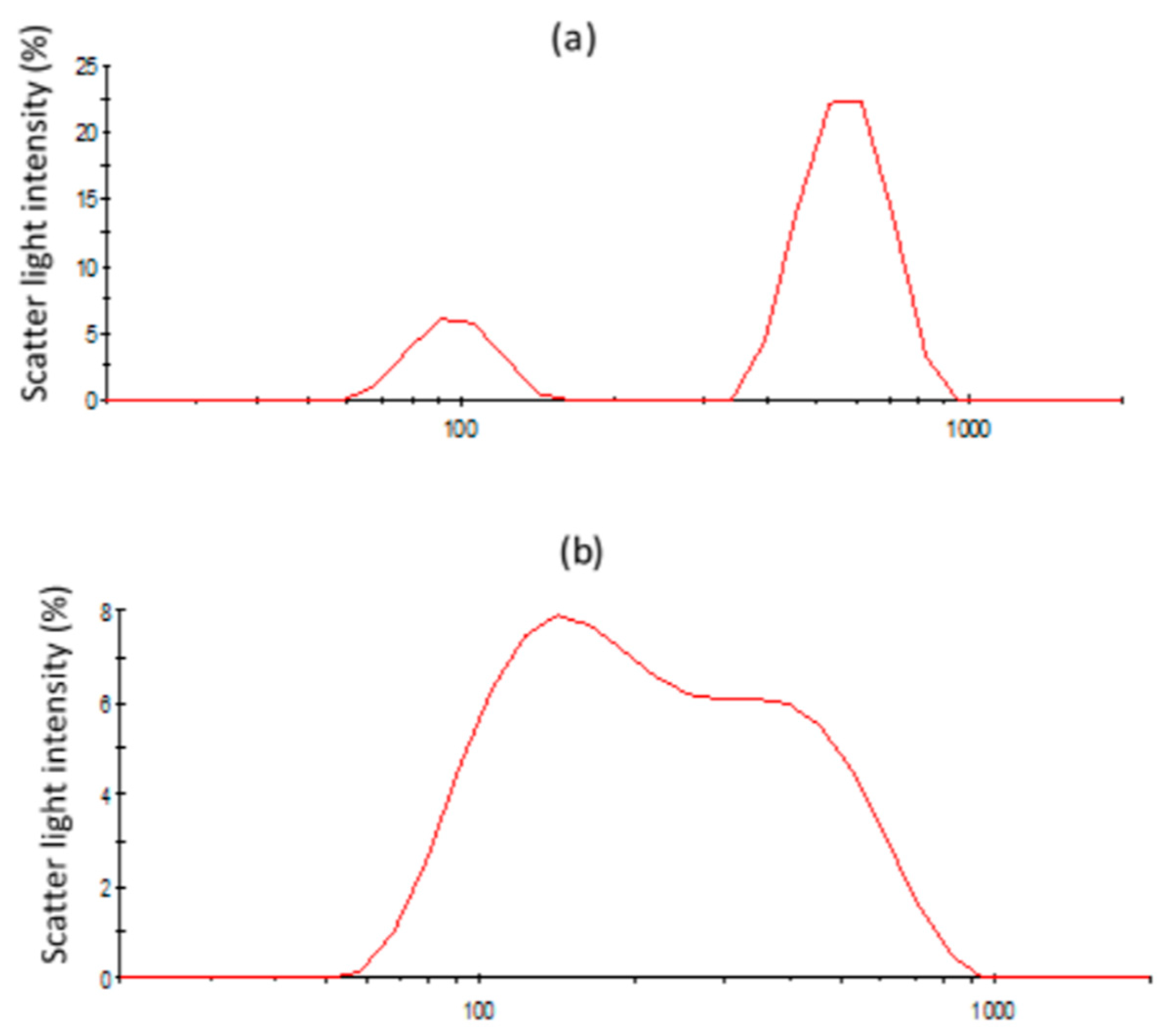
3.3.3. Structural Changes of Emulsions Followed by Light Scattering
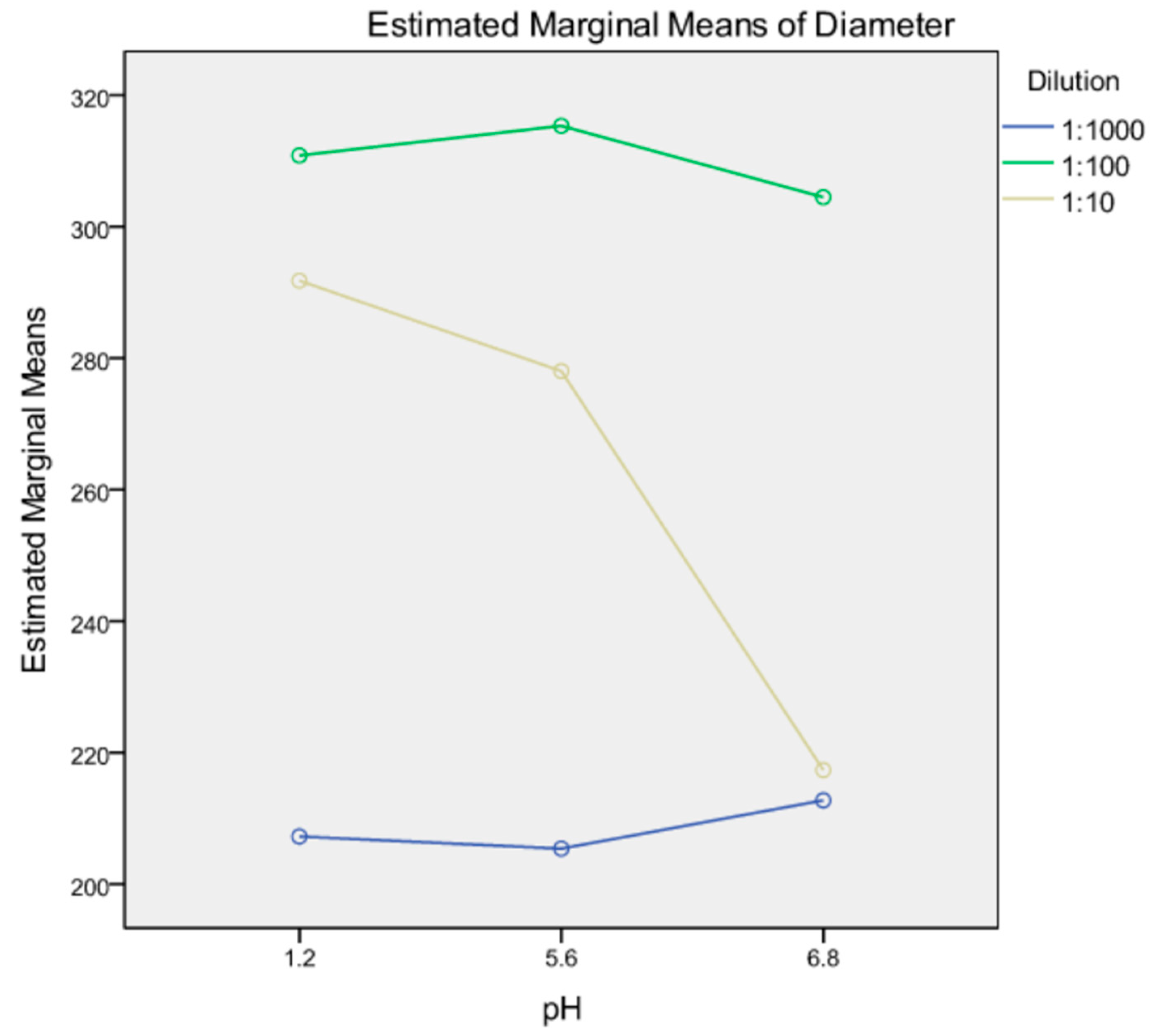
3.3.4. Robustness to Dilution
3.4. Composition of Self-Emulsifying Pellets (SEPs)
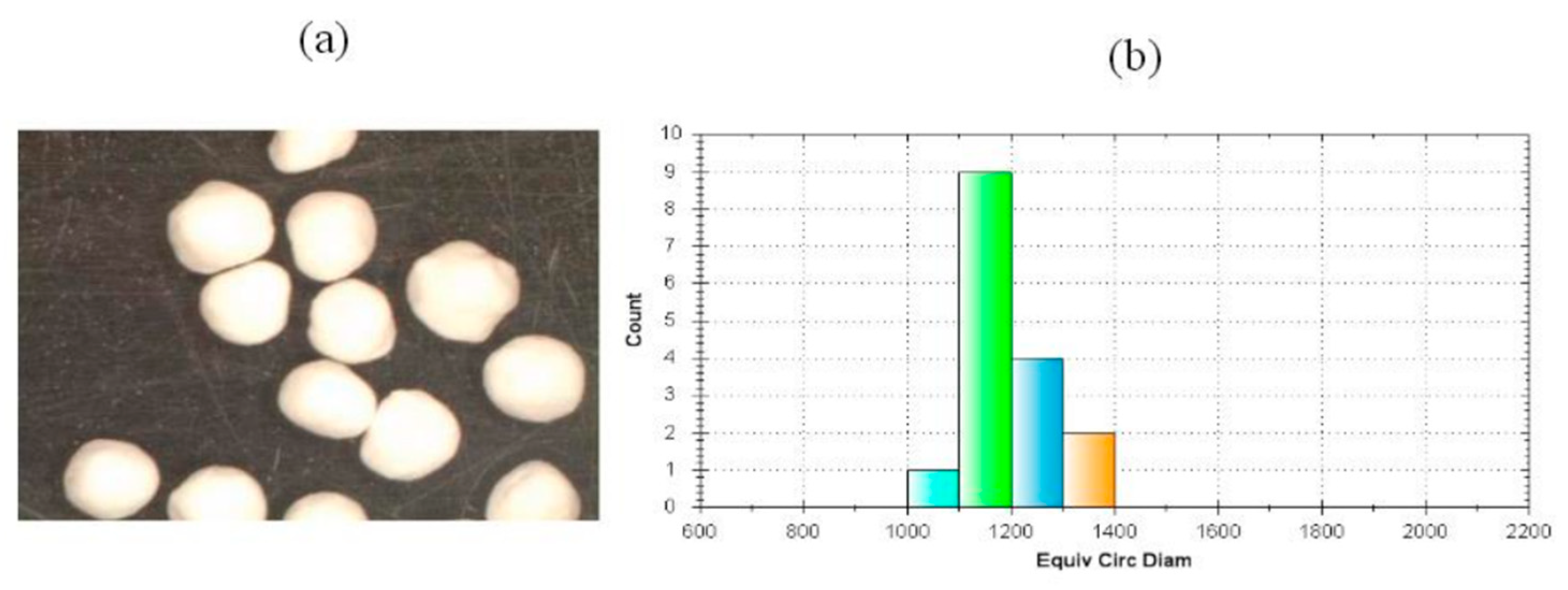
3.5. Particle Size, Shape, and Packing of SEPs
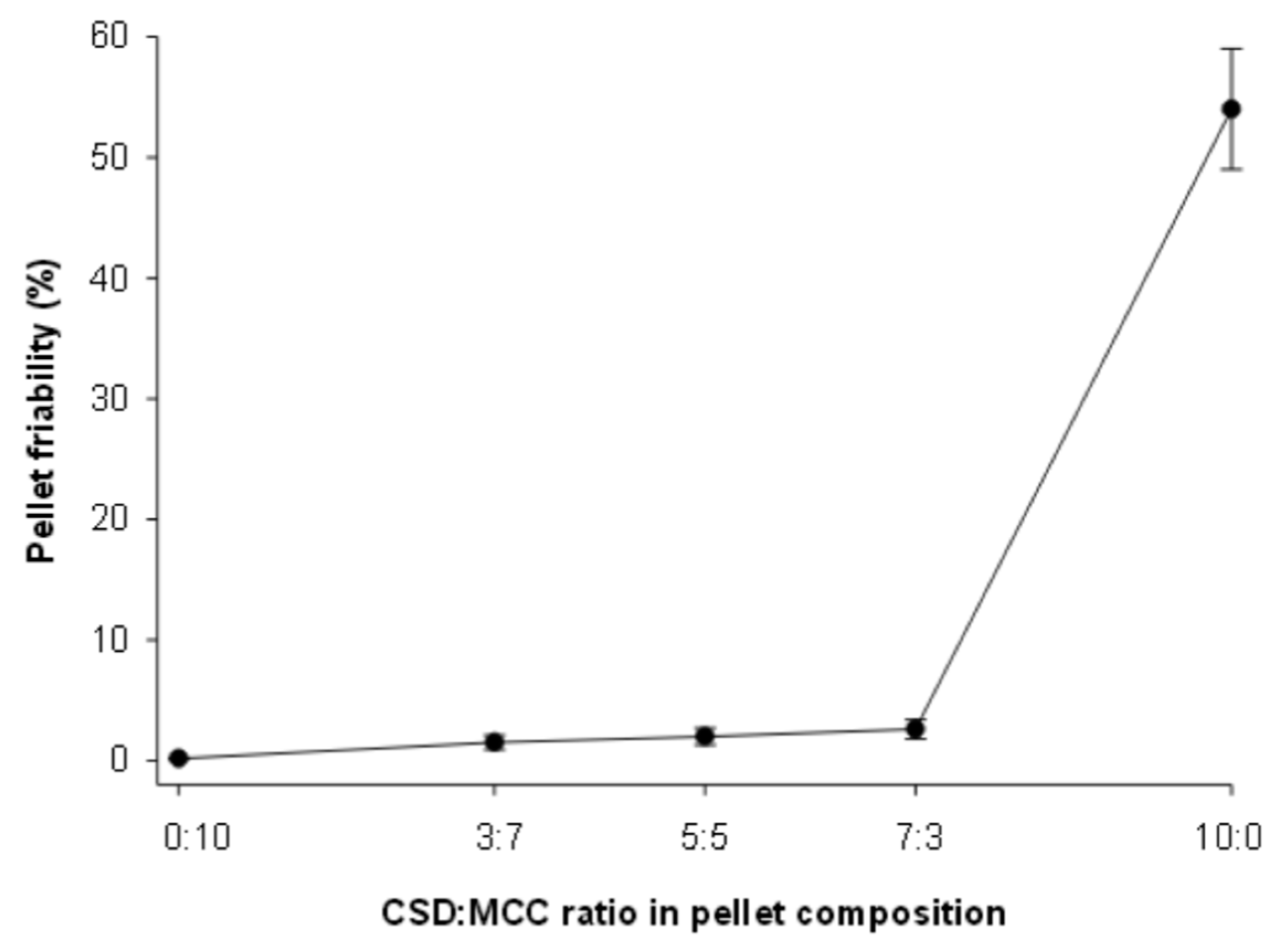
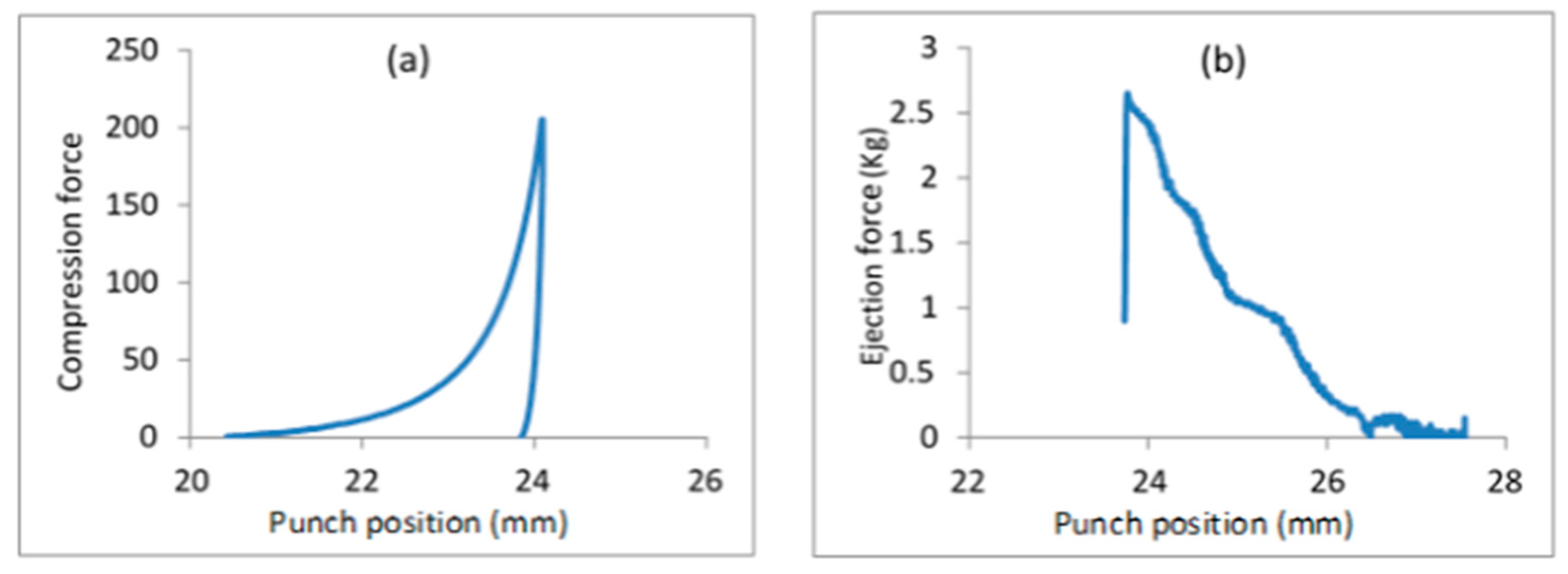
3.6. Friability of SEPs
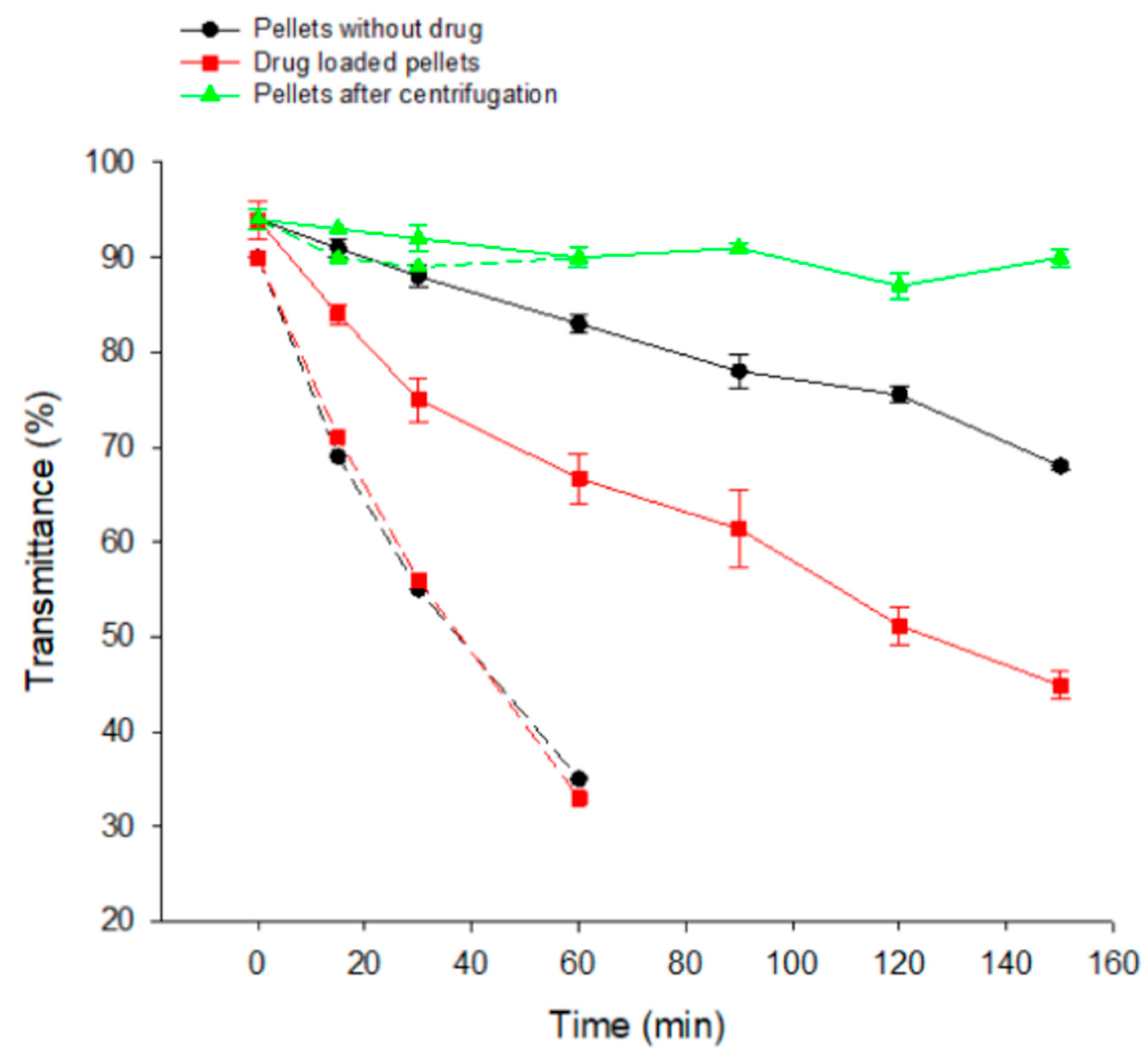
3.7. Reconstitution of Emulsions from SEPs
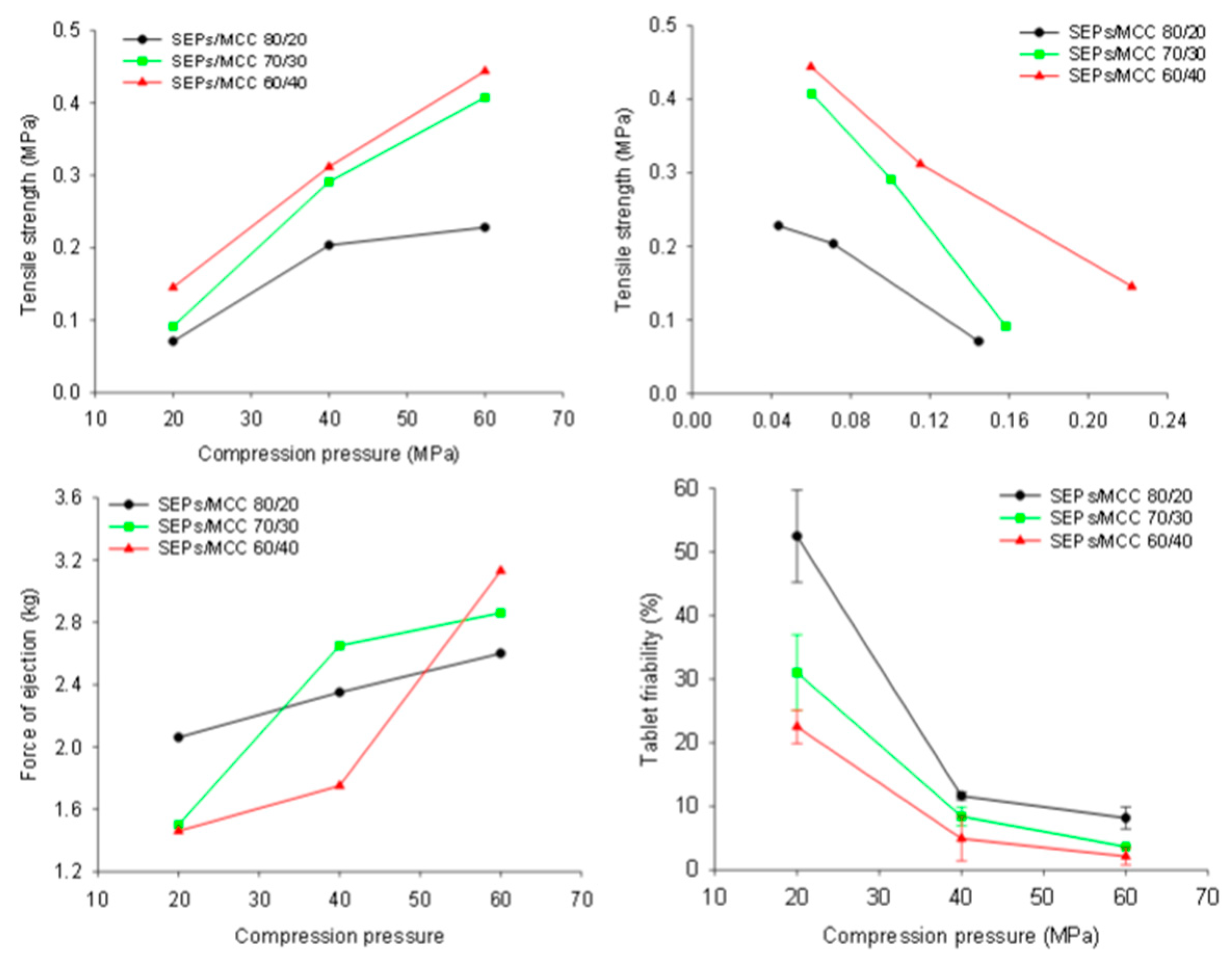
3.8. Self-Emulsifying Tablets
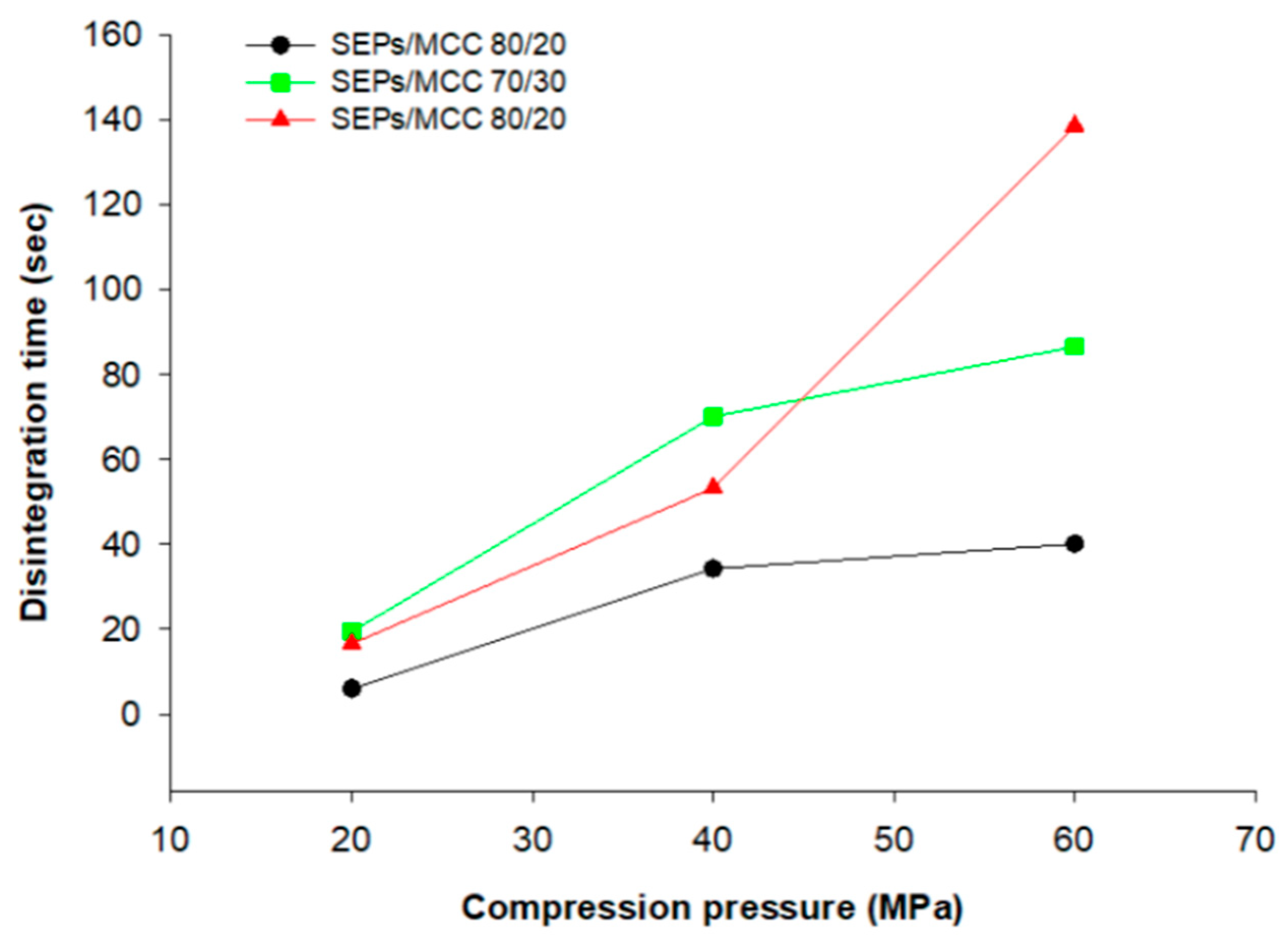
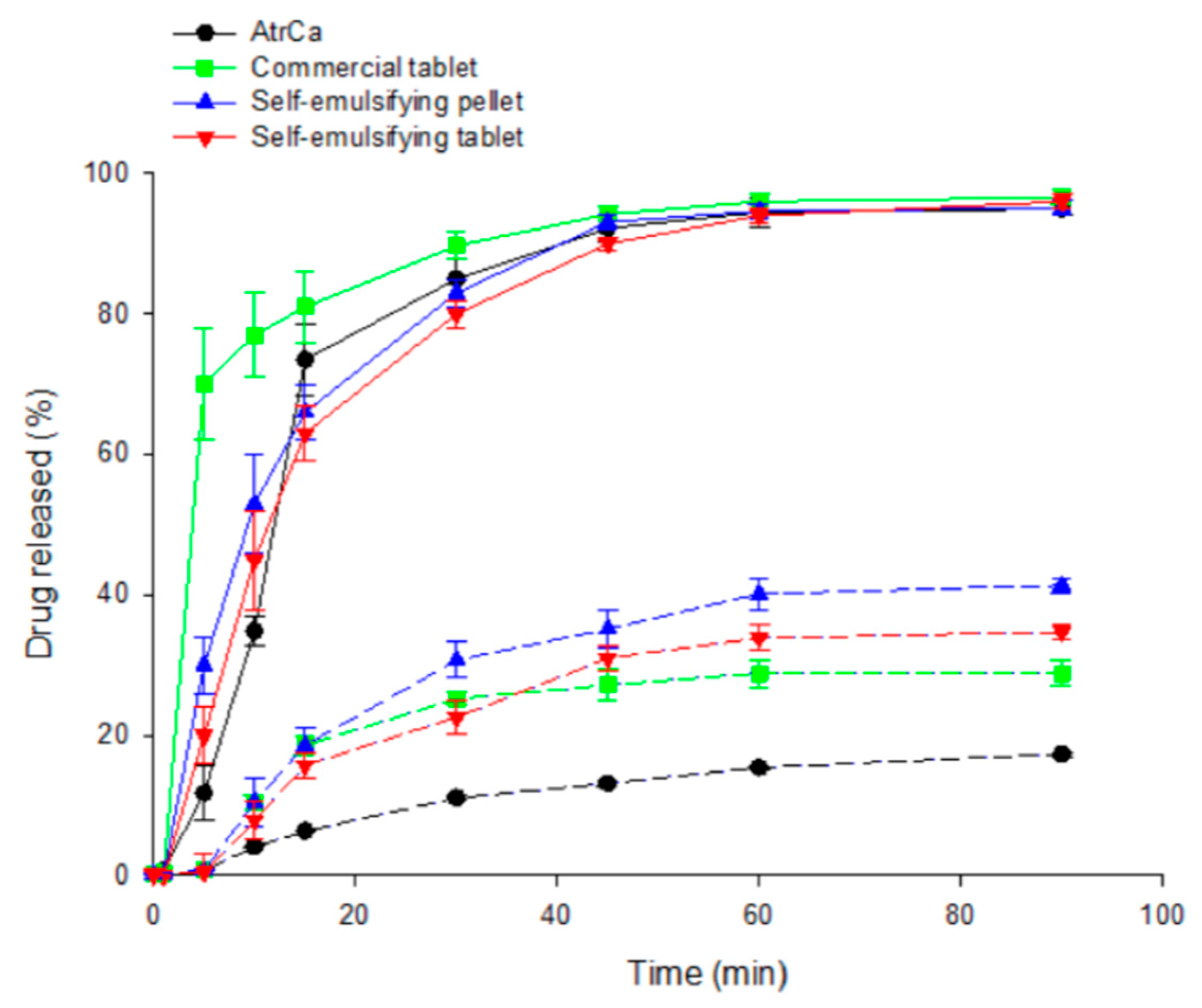
3.9. Disintegration of Tablets into Pellets and In Vitro Release
3.10. Chemical and Physical Stability
3.11. Permeability Study with Caco-2 cell Culture
3.12. Cytotoxicity
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Lea, A.P.; McTavish, D. A Review of its Pharmacology and Therapeutic Potential in the Management of Hyperlipidaemias. Adis Drug 1997, 53, 828–847. [Google Scholar] [CrossRef] [PubMed]
- Athyros, V.G.; Papageorgiou, A.A.; Valasia, V.; Athyrou, V.V.; Demitriadis, D.S.; Anthimos, N.; Pehlivanidis, A.N.; Kontopoulos, A.G. Atorvastatin versus Four Statin–Fibrate Combinations in Patients with Familial Combined Hyperlipidaemia. Eur. J. Prev. Cardiol. 2002, 9, 33–39. [Google Scholar] [CrossRef]
- Lennernäs, H. Clinical pharmacokinetics of atorvastatin. Clin. Pharm. 2003, 42, 1141–1160. [Google Scholar] [CrossRef] [PubMed]
- Lau, Y.Y.; Okochi, H.; Huang, Y.; Benet, L.Z. Pharmacokinetics of atorvastatin and its hydroxy metabolites in rats and the effects of concomitant rifampicin single doses: Relevance of first-pass effect from hepatic uptake transporters, and intestinal and hepatic metabolism. Drug Metab. Dispos. 2006, 34, 1175–1181. [Google Scholar] [CrossRef] [PubMed]
- Paidi, S.K.; Jena, S.K.; Ahuja, B.K.; Devasari, N.; Suresh, S. Preparation, in-vitro and in-vivo evaluation of spray-dried ternary solid dispersion of biopharmaceutics classification system class II model drug. J. Pharm. Pharmacol. 2015, 67, 616–629. [Google Scholar] [CrossRef] [PubMed]
- Kadu, P.J.; Kushare, S.S.; Thacker, D.D.; Gattani, S.G. Enhancement of oral bioavailability of atorvastatin calcium by self-emulsifying drug delivery systems (SEDDS). Pharm. Dev. Technol. 2011, 16, 65–74. [Google Scholar] [CrossRef] [PubMed]
- Karasulu, H.Y.; Gündoğdu, E.; Turgay, T.; Türk, U.Ö.; Apaydın, S.; Şimşir, I.Y.; Yilmaz, C.; Karasulu, E. Development and Optimization of Self-emulsifying Drug Delivery Systems (SEDDS) for Enhanced Dissolution and Permeability of Rosuvastatin. Curr. Drug. Deliv. 2016, 13, 362–370. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.; Zhong, M. Preparation and evaluation of self-microemulsifying drug delivery systems (SMEDDS) containing atorvastatin. J. Pharm Pharmacol. 2006, 58, 1183–1191. [Google Scholar] [CrossRef]
- Govindarajan, R.; Landis, M.; Hancock, B.; Gatlin, L.A.; Suryanarayanan, R.; Shalaev, E.Y. Surface Acidity and Solid-State Compatibility of Excipients with an Acid-Sensitive API: Case Study of Atorvastatin Calcium. AAPS PharmSciTech 2015, 16, 354–363. [Google Scholar] [CrossRef]
- Czajkowska-Kosnik, A.; Szekalska, M.; Amelian, A.; Szymanska, E.; Winnicka, K. Development and Evaluation of Liquid and Solid Self-Emulsifying Drug Delivery Systems for Atorvastatin. Molecules 2015, 20, 21010–21022. [Google Scholar] [CrossRef] [Green Version]
- Gumaste, S.G.; Pawlak, S.A.; Dalrymple, D.M.; Nider, C.J.; Trombetta, L.D.; Serajuddin, A.T.M. Development of Solid SEDDS, IV: Effect of Adsorbed Lipid and Surfactant on Tableting Properties and Surface Structures of Different Silicates. Pharm. Res. 2013, 30, 3170–3185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nikolakakis, I.; Partheniadis, I. Self-Emulsifying Granules and Pellets: Composition and Formation Mechanisms for Instant or Controlled Release. Pharmaceutics 2017, 9, 50. [Google Scholar] [CrossRef] [PubMed]
- Tuleu, C.; Newton, M.; Rose J Euler, D.; Saklatvala, R.; Clarke, A.; Booth, S. Comparative Bioavailability Study in Dogs of a Self-Emulsifying Formulation of Progesterone Presented in a Pellet and Liquid Form Compared with an Aqueous Suspension of Progesterone. J. Pharm. Sci. 2004, 93, 1495–1502. [Google Scholar] [CrossRef] [PubMed]
- Jukkola, A.; Partanen, R.; Xiang, W.; Heino, A.; Rojas, O.J. Food emulsifiers based on milk fat globule membranes and their interactions with calcium and casein phosphoproteins. Food Hydrocoll 2019, 94, 30–37. [Google Scholar] [CrossRef] [Green Version]
- Yildirim, S.T.; Oztop, M.H.; Soyer, Y. Cinnamon oil nanoemulsions by spontaneous emulsification: Formulation, characterization and antimicrobial activity. LWT Food Sci. Technol. 2017, 84, 122–128. [Google Scholar] [CrossRef]
- Wagner, K.G.; Krumme, M.; Schmidt, P.C. Investigation of the pellet-distribution in single tablets via image analysis. Eur. J. Pharm. Biopharm. 1999, 47, 79–85. [Google Scholar] [CrossRef]
- Matsaridou, I.; Barmpalexis, P.; Salis, A.; Nikolakakis, I. The influence of surfactant HLB and oil/surfactant ratio on the formation and properties of Self-emulsifying pellets and microemulsion reconstitution. AAPS PharmSciTech 2012, 13, 1319–1330. [Google Scholar] [CrossRef]
- Gopal, E.S.R. Emulsion Science; Sherman, P., Ed.; Academic Press: New York, NY, USA, 1968; pp. 43–54. [Google Scholar]
- Zetasizer Nanoseries User Mannual MANO 317 Issue 2.2; Malvern Instruments Ltd.: Worcestershire, UK, 2005; p. 16.2.
- Almeida-Prieto, S.; Blanco-Méndez, J.; Francisco JOtero-Espinar, F.J. Image Analysis of the Shape of Granulated Powder Grains. J. Pharm. Sci. 2004, 93, 621–634. [Google Scholar] [CrossRef]
- Fell, J.T.; Newton, J.M. Determination of Tablet Strength by the Diametral-Compression Test. J. Pharm. Sci. 1970, 59, 688–691. [Google Scholar] [CrossRef]
- Choudhary, A.; Rana, A.C.; Aggarwal, G.; Kumar, V.; Zakir, F. Development and characterization of an atorvastatin solid dispersion formulation using skimmed milk for improved oral bioavailability. Acta Pharm. Sin. B 2012, 2, 421–428. [Google Scholar] [CrossRef] [Green Version]
- Başpınar, Y.; Gündoğdu, E.; Köksal, C. Pitavastatin-containing nanoemulsions: Preparation, characterization and in vitro cytotoxicity. J. Drug Deliv. Sci. Technol. 2015, 29, 117–124. [Google Scholar] [CrossRef]
- Bandivadekar, M.; Pancholi, S.; Kaul-Ghanekar, R.; Choudhari, A.; Koppikar, S. Single non-ionic surfactant based selfnanoemulsifying drug delivery systems: Formulation, characterization, cytotoxicity and permeability enhancement study. Drug Dev. Ind. Pharm. 2013, 39, 696–703. [Google Scholar] [CrossRef] [PubMed]
- Dalvadi, H.; Patel, N.; Parmar, K. Systematic development of design of experiments (DoE) optimised self-microemulsifying drug delivery system of Zotepine. J. Microencapsul. 2017, 34, 308–318. [Google Scholar] [CrossRef] [PubMed]
- Quan, D.; Xu, G.; Wu, X. Studies on Preparation and Absolute Bioavailability of a Self-Emulsifying System Containing Puerarin. Chem. Pharm. Bull. 2007, 55, 800–803. [Google Scholar] [CrossRef] [Green Version]
- Zhu, J.; Tang, D.; Feng, L.; Zheng, Z.; Wang, R.; Wu, A.; Duan, T.; He, B.; Zhu, Q. Development of self-microemulsifying drug delivery system for oral bioavailability enhancement of berberine hydrochloride. Drug Dev. Ind. Pharm. 2013, 39, 499–506. [Google Scholar] [CrossRef]
- Pouton, W.C. Formulation of poorly water-soluble drugs for oral administration: Physicochemical and physiological issues and the lipid formulation classification system. Eur. J. Pharm. Sci. 2006, 29, 278–287. [Google Scholar] [CrossRef] [PubMed]
- Magdassi, S.M.; Frenkel, M.; Garti, N. Correlation Between Nature of Emulsifier and Multiple Emulsion Stability. Drug Dev. Ind. Pharm. 1985, 11, 791–798. [Google Scholar] [CrossRef]
- Agrawal, A.G.; Kumar, A.; Gide, P.S. Formulation of solid self-nanoemulsifying drug delivery systems using N-methyl pyrrolidone as cosolvent. Drug Dev. Ind. Pharmac. 2015, 41, 594–604. [Google Scholar] [CrossRef]
- Gershanik, T.; Benita, S. Self-dispersing lipid formulations for improving oral absorption of lipophilic drugs. Eur. J. Pharm. Biopharm. 2000, 50, 179–188. [Google Scholar] [CrossRef]
- Patil, S.S.; Venugopal, E.; Bhat, S.; Mahadik, K.R.; Paradkar, A.R. Microstructural Elucidation of Self-Emulsifying System: Effect of Chemical Structure. Pharm Res. 2012, 29, 2180–2188. [Google Scholar] [CrossRef] [Green Version]
- Maali, A.; Mosavian, M.H. Preparation and Application of Nanoemulsions in the Last Decade (2000–2010). J. Dispers. Sci. Technolog. 2013, 34, 92–105. [Google Scholar] [CrossRef]
- Nikolakakis, I.; Panagopoulou, A.; Salis, A.; Malamataris, S. Relationships between the properties of Self-Emulsifying pellets and of the Emulsions used as massing liquids for their preparation. AAPS PharmSciTech 2015, 16, 129–139. [Google Scholar] [CrossRef] [PubMed]
- Bu, P.; Ji, Y.; Narayanan, S.; Dalrymple, D.; Cheng, X.; Serajuddin, A.T.M. Assessment of cell viability and permeation enhancement in presence of lipid-based self-emulsifying drug delivery systems using Caco-2 cell model: Polysorbate 80 as the surfactant. Eur. J. Pharm. Sci. 2017, 99, 350–360. [Google Scholar] [CrossRef] [PubMed]
- Ditner, C.; Bravo, R.; Imanidis, G.; Kuentz, M. A Systematic Dilution Study of Self-Microemulsifying Drug Delivery Systems in Artificial Intestinal Fluid Using Dynamic Laser Light Backscattering. Drug Dev. Ind. Pharm. 2009, 35, 199–208. [Google Scholar] [CrossRef] [PubMed]
- Petrochenko, P.E.; Pavurala, N.; Wu, Y.; Wong, S.Y.; Parhiz, H.; Chen k Patil, S.M.; Haiou Qu, H.; Buoniconti, P.; Muhammad, A.; Choi, S.; et al. Analytical considerations for measuring the globule size distribution of cyclosporine ophthalmic emulsions. Int. J. Pharm. 2018, 550, 229–239. [Google Scholar]
- Podczeck, F. A novel aid for the preparation of pellets by extrusion/spheronization. Pharm. Technol. Eur. 2008, 20, 26–31. [Google Scholar]
- Nikolakakis, I.; Malamataris, S. Self-Emulsifying Pellets: Relations Between Kinetic Parameters of Drug Release and Emulsion Reconstitution—Influence of Formulation Variables. J. Pharm. Sci. 2014, 103, 1453–1465. [Google Scholar] [CrossRef]
- Kapsidou, T.; Nikolakakis, I.; Malamataris, S. Agglomeration state and migration of drugs in wet granulations during drying. Int. J. Pharm. 2001, 227, 97–112. [Google Scholar] [CrossRef]
- Ettlinger, M.; Ferch, H.; Mathias, J. Reviews and Trends: Grundlagen und Anwendungen einer durch Flammenhydrolyse gewonnenen Kieselsäure, 25. Mitt. Zur Adsorption an der Oberfläche einer flammenhydrolytisch gewonnenen Kieselsäure. Arch. Pharm. 1987, 320, 1–15. (In German) [Google Scholar] [CrossRef]
- Nasr, A.; Ahmed Gardouh, A.; Ghora, M. Novel Solid Self-Nanoemulsifying Drug Delivery System (S-SNEDDS) for Oral Delivery of Olmesartan Medoxomil: Design, Formulation, Pharmacokinetic and Bioavailability Evaluation. Pharmaceutics 2016, 8, 20. [Google Scholar] [CrossRef]
- Amiri, A.; Øye, G.; Sjöblom, J. Influence of pH, high salinity and particle concentration on stability and rheological properties of aqueous suspensions of fumed silica. Colloids Surf. A Physicochem. Eng. Asp. 2009, 349, 43–54. [Google Scholar] [CrossRef]
- Sun, C.C. Dependence of ejection force on tableting speed—A compaction simulation study. Powder Technol. 2015, 279, 123–126. [Google Scholar] [CrossRef]
- Gumaste, S.G.; Damon, M.; Dalrymple, D.M.; Serajuddin, A.T.M. Development of Solid SEDDS, V: Compaction and Drug Release Properties of Tablets Prepared by Adsorbing Lipid-Based Formulations onto Neusilin® US2. Pharm. Res. 2013, 30, 3186–3199. [Google Scholar] [CrossRef] [PubMed]
- Hamman, M.A.; Bruce, M.A.; Haehner-Daniels, B.D.; Hall, S.D. The Effect of Rifampin Administration on the Disposition of Fexofenadine. Clin. Pharm. Ther. 2001, 69, 114–121. [Google Scholar] [CrossRef] [PubMed]
- Hauri, H.P.; Sterchi, E.E.; Bienz, D.; Fransen, J.A.; Marxer, A. Expression and Intracellular Transport of Microvillus Membrane Hydrolases in Human Intestinal Epithelial Cells. J. Cell Biol. 1985, 101, 838–851. [Google Scholar] [CrossRef] [PubMed]
- Hilers, A.R.; Conradi, R.A.; Burton, P.S. Caco-2 Cell Monolayers as a Model for Drug Transport Across the Intestinal Mucosa. Pharm. Res. 1990, 7, 902–910. [Google Scholar] [CrossRef]
- Gundogdu, E.; Mangas-Sanjuan, V.; Gonzalez, A.I.; Bermejo, M.; Karasulu, E. In vitro-in situ permeability and dissolution of fexofenadine with kinetic modeling in presence of sodium dodecyl sulfate. Eur. J. Drug Metab. Pharmacokinet. 2012, 37, 65–75. [Google Scholar] [CrossRef]
- González-Alvarez, I.; Fernández-Teruel, C.; Garrigues, T.M.; Casabo, V.G.; Ruiz-García, A.; Bermejo, M. Kinetic Modelling of Passive Transport and Active Efflux of a Fluoroquinolone Across Caco–2 Cell Using a Compartmental Approach In NONMEM. Xenobiotica 2005, 35, 1067–1088. [Google Scholar] [CrossRef]
- Okyar, A. P-glikoprotein ve P-glikoproteinin İlaç Farmakokinetiğindeki Rolü. Türk Farm. Derneği Bülteni 2005, 83, 18–22. [Google Scholar]
- Arturson, P.; Palm, K.; Luthman, K. Caco-2 Cell Monolayers in Experimental and Theoretical Predictions of Drug Transport. Adv. Drug Del. Rev. 2001, 46, 27–43. [Google Scholar]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ingredient | Solubility (mg/mL) (±SD) |
|---|---|
| Corn oil | 7.71 ± 0.05 |
| Oleic acid | 27.40 ± 0.03 |
| Refined castor oil | 1.53 ± 0.02 |
| Labrofil M 2125 | 4.47 ± 0.04 |
| Labrafil 1944 | 0.01 ± 0.04 |
| Tween 20 | 263.5 ± 0.09 |
| Tween 80 | 13.50 ± 0.02 |
| Span 80 | 47.47 ± 0.03 |
| PEG 200 | 45.98 ± 0.03 |
| Transcutol | 6.74 ± 0.04 |
| Labrasol | 17.30 ± 0.18 |
| Brij 30v | 44.73 ± 0.21 |
| Brij 92v | 30.19 ± 0.04 |
| Brij 96v | 3.38 ± 0.03 |
| Ethanol | 0.04 ± 0.02 |
| Isopropyl alcohol | 4.71 ± 0.06 |
| N-methyl pyrrolidone | 110.55 ± 0.03 |
| Distilled water | 0.01 ± 0.05 |
| pH: 1.2 | 0.004 ± 0.02 |
| pH: 4.6 | 0.02 ± 0.01 |
| pH: 6.8 | 0.602 ± 0.03 |
| Ingredient | Composition |
|---|---|
| Drug | 1.2 (2.7%) |
| Aerosil 200 | 14.0 (31.1%) |
| Avicel 101 | 6.0 (13.3%) |
| Oleic acid | 14.4 (32.0%) |
| Tween 20 | 5.7 (12.6%) |
| Span 80 | 2.8 (6.2%) |
| N-methyl pyrrolidone | 0.96 (2.1%) |
| Total weight | 45.0 (100%) |
| Self-Emulsifying Pellets (SEPs) | Avicel 101 | Compression (MPa) |
|---|---|---|
| 20 | 80 | 20 |
| 20 | 80 | 40 |
| 20 | 80 | 60 |
| 30 | 70 | 20 |
| 30 | 70 | 40 |
| 30 | 70 | 60 |
| 40 | 60 | 20 |
| 40 | 60 | 40 |
| 40 | 60 | 60 |
| Property | Non-Sonicated | Sonicated |
|---|---|---|
| Droplet size | 1370 ± 23 | 320.7 ± 15 |
| Viscosity (cP) | 249.5 | 68.5 |
| Zeta potential (mV) | −7.05 ± 0.48 | −7.98 ± 0.35 |
| Dispersion Medium | Dilution Level | Diameter (nm) | Coefficient of Variation (CV%) |
|---|---|---|---|
| pH 1.2 | 1:10 | 288.6 ± 1.5 | 19.3 |
| pH 1.2 | 1:100 | 306.7 ± 1.0 | |
| pH 1.2 | 1:1000 | 209.5 ± 1.5 | |
| pH 1.2 | 1:1000/24 h | 210.9 ± 1.0 | |
| pH 6.8 | 1:10 | 215.7 ± 1.5 | 21.6 |
| pH 6.8 | 1:100 | 307.0 ± 1.3 | |
| pH 6.8 | 1:1000 | 214.5 ± 1.0 | |
| pH 6.8 | 1:1000/24 h | 285.9 ± 1.3 | |
| Deionised water | 1:10 | 273.1 ± 2.6 | 22.8 |
| Deionised water | 1:100 | 320.7 ± 1.5 | |
| Deionised water | 1:1000 | 200.8 ±2.3 | |
| Deionised water | 1:1000/24 h | 198.4 ± 1.8 |
| Median Diameter (μm) | 1065 |
| Shape index (eR) | 0.469 |
| Circularity | 0.832 |
| Pycnometric density | 1.31 ± 0.01 |
| Bulk density | 0.72 ± 0.04 |
| Tap density | 0.77 ± 0.02 |
| Hausner ratio | 1.07 ± 0.01 |
| Carr’s index | 6.46 ± 0.61 |
| Friability (%) | 1.3 ± 0.05 |
| Time 0 | Three Months | Six Months | ||||
|---|---|---|---|---|---|---|
| 25 °C/60% | 40 °C/75% | 25 °C/60% | 40 °C/75% | 25 °C/60% | 40 °C/75% | |
| Drug (%) | 106 ± 1.53 | 103 ± 1.35 | 104 ± 2.00 | 102 ± 1.87 | 102 ± 1.99 | 101 ± 1.47 |
| Apparent Permeability Coefficient | Pefflux | Cytotoxicity | ||
|---|---|---|---|---|
| A→B | B→A | |||
| AtrCa | 8.01 × 10−5 ± 5.56 × 10−6 | 1.59 × 10−4 ± 4.03 × 10−5 | 0.503 0.12 | 72% |
| Self-emulsifying pellets | 6.16 × 10−4 ± 1.23 × 10−5 | 5.85 × 10−4 ± 5.67 × 10−6 | 1.046 0.02 | 88.9% |
| Commercial Tablet | 4.87 × 10−4 ± 8.3 × 10−5 | 6.79 × 10−4 ± 5.37 × 10−5 | 0.718 0.15 | 89.9% |
| Apparent Permeability Coefficient | Pefflux | ||
|---|---|---|---|
| A→B | B→A | ||
| AtrCa - SEP | 0.005 | 0.000 | 0.002 |
| AtrCa–Comm. tabl. | 0.000 | 0.000 | 0.096 |
| SEP–Comm. Tabl. | 0.026 | 0.051 | 0.020 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Diril, M.; Karasulu, Y.; Toskas, M.; Nikolakakis, I. Development and Permeability Testing of Self-Emulsifying Atorvastatin Calcium Pellets and Tablets of Compressed Pellets. Processes 2019, 7, 365. https://doi.org/10.3390/pr7060365
Diril M, Karasulu Y, Toskas M, Nikolakakis I. Development and Permeability Testing of Self-Emulsifying Atorvastatin Calcium Pellets and Tablets of Compressed Pellets. Processes. 2019; 7(6):365. https://doi.org/10.3390/pr7060365
Chicago/Turabian StyleDiril, Mine, Yesim Karasulu, Miltiadis Toskas, and Ioannis Nikolakakis. 2019. "Development and Permeability Testing of Self-Emulsifying Atorvastatin Calcium Pellets and Tablets of Compressed Pellets" Processes 7, no. 6: 365. https://doi.org/10.3390/pr7060365