Nitroxide-Mediated Copolymerization of Itaconate Esters with Styrene


Abstract
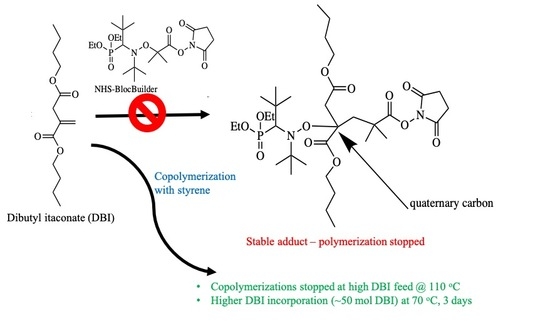
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. DBI/S Copolymerization
2.3. Chain Extension Experiments
2.4. Characterization
3. Results and Discussion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Robert, T.; Friebel, S. Itaconic acid—A versatile building block for renewable polyesters with enhanced functionality. Green Chem. 2016, 18, 2922–2934. [Google Scholar] [CrossRef]
- Tate, B.E. Polymerization of itaconic acid and derivatives. Adv. Poly. Sci. 1967, 5, 214–232. [Google Scholar]
- Velada, J.; Hernáez, E.; Cesteros, L.C.; Katime, I. Study of the thermal degradation of several poly (monoalkylaryl itaconates). Polym. Degrad. Stab. 1996, 52, 273–282. [Google Scholar] [CrossRef]
- Calam, C.T.; Oxford, A.E.; Raistrick, H. Studies in the biochemistry of micro-organisms: Itaconic acid, a metabolic product of a strain of Aspergillus terreus Thom. Biochem. J. 1939, 33, 1488. [Google Scholar] [CrossRef] [PubMed]
- Willke, T.; Vorlop, K.-D. Biotechnological production of itaconic acid. Appl. Microbiol. Biotechnol. 2001, 56, 289–295. [Google Scholar] [CrossRef]
- Willke, T.; Vorlop, K.-D. Industrial bioconversion of renewable resources as an alternative to conventional chemistry. Appl. Microbiol. Biotechnol. 2004, 66, 131–142. [Google Scholar] [CrossRef]
- Dai, J.; Liu, X.; Ma, S.; Wang, J.; Shen, X.; You, S.; Zhu, J. Soybean oil-based UV-curable coatings strengthened by crosslink agent derived from itaconic acid together with 2-hydroxyethyl methacrylate phosphate. Prog. Org. Coat. 2016, 97, 210–215. [Google Scholar] [CrossRef]
- Katime, I.; Rodríguez, E. Absorption of metal ions and swelling properties of poly (acrylic acid-co-itaconic acid) hydrogels. J. Macromol. Sci. Part A Pure Appl. Chem. 2001, 38, 543–558. [Google Scholar] [CrossRef]
- Werpy, T.; Petersen, G.; Aden, A.; Bozell, J.; Holladay, J.; White, J.; Manheim, A. Top Value Added Chemicals from Biomass Vol. I—Results of Screening for Potential Candidates from Sugars and Synthesis Gas; Report DOE/GO-102004-1992; U.S. Department of Energy, Office of Scientific and Technical Information: Washington, DC, USA, 2004. Available online: https://www.nrel.gov/docs/fy04osti/35523.pdf (accessed on 30 April 2019).
- Klement, T.; Büchs, J. Itaconic Acid—A Biotechnological Process in Change. Bioresour. Technol. 2013, 135, 422–431. [Google Scholar] [CrossRef]
- Marvel, C.S.; Shepherd, T.H. Polymerization Reactions of Itaconic Acid and Some of Its Derivatives. J. Org. Chem. 1959, 24, 599–605. [Google Scholar] [CrossRef]
- Takasu, A.; Ito, M.; Inai, Y.; Hirabayashi, T.; Nishimura, Y. Synthesis of biodegradable polyesters by ring-opening copolymerization of cyclic anhydrides containing a double bond with 1,2-epoxybutane and one-pot preparation of the itaconic acid-based polymeric network. Polym. J. 1999, 31, 961–969. [Google Scholar] [CrossRef]
- Tang, T.; Moyori, T.; Takasu, A. Isomerization-free polycondensations of cyclic anhydrides with diols and preparation of polyester gels containing cis or trans carbon double bonds via photo-cross-linking and isomerization in the gels. Macromolecules 2013, 46, 5464–5472. [Google Scholar] [CrossRef]
- Farmer, T.; Castle, R.; Clark, J.; Macquarrie, D. Synthesis of unsaturated polyester resins from various bio-derived platform molecules. Int. J. Mol. Sci. 2015, 16, 14912–14932. [Google Scholar] [CrossRef] [PubMed]
- Winkler, M.; Lacerda, T.M.; Mack, F.; Meier, M.A.R. Renewable polymers from itaconic acid by polycondensation and ring-opening-metathesis polymerization. Macromolecules 2015, 48, 1398–1403. [Google Scholar] [CrossRef]
- Bai, Y.; De bruyn, M.; Clark, J.H.; Dodson, J.R.; Farmer, T.J.; Honore, M.; Ingram, I.D.V.; Naguib, M.; Whitwood, A.C.; North, M. Ring opening metathesis polymerization of a new bio-derived monomer from itaconic anhydride and furfuryl alcohol. Green Chem. 2016, 18, 3945–3948. [Google Scholar] [CrossRef]
- Lv, A.; Li, Z.-L.; Du, F.-S.; Li, Z.-C. Synthesis, functionalization, and controlled degradation of high molecular weight polyester from itaconic acid via ADMET polymerization. Macromolecules 2014, 47, 7707–7716. [Google Scholar] [CrossRef]
- Nagai, S.; Yoshida, K. Polymerization of itaconic acid derivatives. Part III: Rate of polymerization of dialkyl itaconates. Kobunshi Kagaku 1960, 17, 79–82. [Google Scholar] [CrossRef]
- Braun, V.D.; Ahn, T.-O. Copolymerisation von Itaconsauredialkylestern mit Styrol. Kolloid Z. 1963, 188, 1–4. [Google Scholar] [CrossRef]
- Sato, T.; Morita, N.; Tanaka, H.; Ota, T. Solvent effect on the radical polymerization of di-n-butyl itaconate. J. Polym. Sci. A Polym. Chem. 1989, 27, 2497–2508. [Google Scholar] [CrossRef]
- Otsu, T.; Watanabe, H. Radical polymerization reactivity of dialkyl itaconates and characterization of their polymers. Eur. Polym. J. 1993, 29, 167–174. [Google Scholar] [CrossRef]
- Fernandez-Garcia, M.; Madruga, E.L. Glass transition in dimethyl and di-n-butyl poly(itaconate ester)s and their copolymers with methyl methacrylate. Polymer 1997, 38, 1367–1371. [Google Scholar] [CrossRef]
- Madruga, E.L.; Fernandez-Garcia, M. Free-radical homopolymerization and copolymerization of di-n-butyl itaconate. Polymer 1994, 35, 4437–4442. [Google Scholar] [CrossRef]
- Veličković, J.; Filipović, J.; Djakov, D.P. The synthesis and characterization of poly(itaconic) acid. Polym. Bull. 1994, 32, 169–172. [Google Scholar] [CrossRef]
- Hirano, T.; Takeyoshi, R.; Seno, M.; Sato, T. Chain-transfer reaction in the radical polymerization of di-n-butyl itaconate at high temperatures. J. Polym. Sci. A Polym. Chem. 2002, 40, 2415–2426. [Google Scholar] [CrossRef]
- Hirano, T.; Higashi, K.; Seno, M.; Sato, T. Radical polymerization of di-n-butyl itaconate in the presence of Lewis acids. Eur. Polym. J. 2003, 39, 1801–1808. [Google Scholar] [CrossRef]
- Hirano, T.; Higashi, K.; Seno, M.; Sato, T. Reaction control in radical polymerization of di-n-butyl itaconate utilizing a hydrogen-bonding interaction. J. Polym. Sci. A Polym. Chem. 2004, 42, 4895–4905. [Google Scholar] [CrossRef]
- Rangel-Rangel, E.; Torres, C.; Rincon, L.; Loteich-Khatib, S.; Lopez-Carrasquero, F. Copolymerizations of long side chain di n-alkyl itaconates and methyl n-alkyl itaconates with styrene: Determination of monomers reactivity ratios by NMR. Rev. Latinoam. Metal. Mater. 2012, 32, 79–88. [Google Scholar]
- Lopez-Carrasqero, F.; Rangel-Rangel, E.; Cardenas, M.; Torres, C.; Dugarte, N.; Laredo, E. Copolymers of long-side-chain di-n-alkyl itaconates or methyl m-alkyl itaconates with styrene: Synthesis, characterization, and thermal properties. Polym. Bull. 2013, 70, 131–146. [Google Scholar] [CrossRef]
- Sarkar, P.; Bhowmick, A.K. Green approach toward sustainable polymer: Synthesis and characterization of poly(myrcene-co-dibutyl itaconate). ACS Sustain. Chem. Eng. 2016, 4, 2129–2141. [Google Scholar] [CrossRef]
- Lei, W.; Russell, T.P.; Hu, L.; Zhou, X.; Qiao, H.; Wang, W.; Wang, R.; Zhang, L. Pendant Chain effect on the synthesis, characterization, and structure–property relations of poly (di-n-alkyl itaconate-co-isoprene) biobased elastomers. ACS Sustain. Chem. Eng. 2017, 5, 5214–5223. [Google Scholar] [CrossRef]
- Szablan, Z.; Toy, A.A.; Terrenoire, A.; Davis, T.P.; Stenzel, M.H.; Müller, A.H.E.; Barner-Kowollik, C. Living free-radical polymerization of sterically hindered monomers: Improving the understanding of 1,1 di-substituted monomer systems. J. Polym. Sci. A Polym. Chem. 2006, 44, 3692–3710. [Google Scholar] [CrossRef]
- Satoh, K.; Lee, D.-H.; Nagai, K.; Kamigaito, M. Precision Synthesis of Bio-Based Acrylic Thermoplastic Elastomer by RAFT Polymerization of Itaconic Acid Derivatives. Macromol. Rapid Commun. 2014, 35, 161–167. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Garcia, M.; Fernandez-Sanz, M.; De la Fuente, J.L.; Madruga, E.L. Atom-transfer radical polymerization of dimethyl itaconate. Macromol. Chem. Phys. 2001, 202, 1213–1218. [Google Scholar] [CrossRef]
- Okada, S.; Matyjaszewski, K. Synthesis of bio-based poly(N-phenylitaconimide) by atom transfer radical polymerization. J. Polym. Sci. A Polym. Chem. 2015, 53, 822–827. [Google Scholar] [CrossRef]
- Inciarte, H.; Orozco, M.; Fuenmayor, M.; López-Carrasquero, F.; Oliva, H. Comb-like copolymers of n-alkyl monoitaconates and styrene. e-Polymers 2006, 6. [Google Scholar] [CrossRef]
- Nagai, S. The polymerization and polymers of itaconic acid derivatives. VI. The polymerization and copolymerization of itaconic anhydride. Bull. Chem. Soc. Jpn. 1964, 37, 369–373. [Google Scholar] [CrossRef]
- Wallach, J.A.; Huang, S.J. Copolymers of itaconic anhydride and methacrylate-terminated poly(lactic acid) macromonomers. Biomacromolecules 2000, 1, 174–179. [Google Scholar] [CrossRef]
- Shang, S.; Huang, S.J.; Weiss, R.A. Synthesis and characterization of itaconic anhydride and stearyl methacrylate copolymers. Polymer 2009, 50, 3119–3127. [Google Scholar] [CrossRef]
- Javakhishvili, I.; Kasama, T.; Jankova, K.; Hvilsted, S. RAFT copolymerization of itaconic anhydride and 2-methoxyethyl acrylate: A multi-functional scaffold for preparation of “clickable” gold nanoparticles. Chem. Commun. 2013, 49, 4803–4805. [Google Scholar] [CrossRef]
- Nicolas, J.; Guillaneuf, Y.; Lefay, C.; Bertin, D.; Gigmes, D.; Charleux, B. Nitroxide mediated polymerization. Prog. Polym. Sci. 2013, 38, 63–235. [Google Scholar] [CrossRef]
- Willcock, H.; O’Reilly, R.K. End group removal and modification of RAFT polymers. Polym. Chem. 2010, 1, 149–157. [Google Scholar] [CrossRef]
- Su, X.; Jessop, P.G.; Cunningham, M.F. ATRP catalyst removal and ligand recycling using CO2-switchable materials. Macromolecules 2018, 51, 8156–8164. [Google Scholar] [CrossRef]
- Wang, Y.; Lorandi, F.; Fantin, M.; Chmielarz, P.; Isse, A.A.; Gennaro, A.; Matyjaszewski, K. Miniemulsion ARGET ATRP via interfacial and ion-pair catalysis: From ppm to ppb of residual copper. Macromolecules 2017, 50, 8417–8425. [Google Scholar] [CrossRef]
- Vinas, J.; Chagneux, N.; Gigmes, D.; Trimaille, T.; Favier, A.; Bertin, D. SG1-based alkoxyamine bearing a N-succinimidyl ester: A versatile tool for advanced polymer synthesis. Polymer 2008, 49, 3639–3647. [Google Scholar] [CrossRef]
- Szablan, Z.; Stenzel, M.H.; Davis, T.P.; Barner, L.; Barner-Kowollik, C. Depropagation kinetics of sterically demanding monomers: A pulsed laser size exclusion chromatography study. Macromolecules 2005, 38, 5944–5954. [Google Scholar] [CrossRef]
- Buback, M.; Gilbert, R.G.; Hutchinson, R.A.; Klumperman, B.; Kuchta, F.D.; Manders, B.G.; O’Driscoll, K.F.; Russell, G.T.; Schweer, J. Critically evaluated rate coefficients for free-radical polymerization, 1. Propagation rate coefficient for styrene. Macromol. Chem. Phys. 1995, 196, 3267–3280. [Google Scholar] [CrossRef]
- Kassi, E.; Loizou, E.; Porcar, L.; Patrickios, C.S. Di(n-butyl) itaconate end-functionalized polymers: Synthesis by group transfer polymerization and solution characterization. Eur. Polym. J. 2011, 47, 816–822. [Google Scholar] [CrossRef]
- Kassi, E.; Constantinou, M.S.; Patrickios, C.S. Group transfer polymerization of bio-based monomers. Eur. Polym. J. 2013, 49, 761–767. [Google Scholar] [CrossRef]
- Tomić, S.L.; Filipović, J.M.; Velicković, J.S.; Katsikas, L.; Popović, I.G. The polymerisation kinetics of lower dialkyl itaconates. Macromol. Chem. Phys. 1999, 200, 2421–2427. [Google Scholar] [CrossRef]
- Benoit, D.; Grimaldi, S.; Robin, S.; Finet, J.P.; Tordo, P.; Gnanou, Y. Kinetics and mechanism of controlled free-radical polymerization of styrene and n-butyl acrylate in the presence of an acyclic-phosphonylated nitroxide. J. Am. Chem. Soc. 2000, 122, 5929–5939. [Google Scholar] [CrossRef]
- Fukuda, T.; Ma, Y.-D.; Inagaki, H. Free radical copolymerization. 3. Determination of rate constants of propagation and termination for styrene/methyl methacrylate system. A critical test of terminal-model kinetics. Macromolecules 1985, 18, 17–26. [Google Scholar] [CrossRef]
- Fischer, H. The persistent radical effect in controlled radical polymerizations. J. Polym. Sci. A Polym. Chem. 1999, 37, 1885–1901. [Google Scholar] [CrossRef]
- Charleux, B.; Nicolas, J.; Guerret, O. Theoretical expression of the average activation-deactivation equilibrium constant in controlled/living free-radical copolymerization operating via reversible termination. Application to a strongly improved control in nitroxide-mediated polymerization of methyl methacrylate. Macromolecules 2005, 38, 5484–5492. [Google Scholar]
- Ma, Y.D.; Sung, K.S.; Tsujii, Y.; Fukuda, T. Free-radical copolymerization of styrene and diethyl fumarate. Penultimate-unit effects on both propagation and termination processes. Macromolecules 2001, 34, 4749–4756. [Google Scholar] [CrossRef]
- Lessard, B.; Marić, M. Effect of acrylic acid neutralization on ‘livingness’ of poly[styrene-ran-(acrylic acid)] macro-initiators for nitroxide-mediated polymerization of styrene. Polym. Int. 2008, 57, 1141–1151. [Google Scholar] [CrossRef]
- Buback, M.; Egorov, M.; Junkers, T.; Panchenko, E. Termination kinetics of dibutyl itaconate free-radical polymerization studied via the SP–PLP–ESR technique. Macromol. Chem. Phys. 2005, 206, 333–341. [Google Scholar] [CrossRef]
- Buback, M.; Kuchta, F.D. Termination kinetics of free-radical polymerization of styrene over an extended temperature and pressure range. Macromol. Chem. Phys. 1997, 198, 1455–1480. [Google Scholar] [CrossRef]
- Yee, L.H.; Heuts, J.A.P.; Davis, T.P. Copolymerization propagation kinetics of dimethyl itaconate and styrene: Strong entropic contributions to the penultimate unit effect. Macromolecules 2001, 34, 3581–3586. [Google Scholar] [CrossRef]
- Szablan, Z.; Toy, A.A.; Davis, T.P.; Hao, X.; Stenzel, M.H.; Barner-Kowollik, C. Reversible addition fragmentation chain transfer polymerization of sterically hindered monomers: Toward well-defined rod/coil architectures. J. Polym. Sci. A Polym. Chem. 2004, 42, 2432–2443. [Google Scholar] [CrossRef]
- Marque, S.; Le Mercier, C.; Tordo, P.; Fischer, H. Factors influencing the C–O–bond homolysis of trialkylhydroxylamines. Macromolecules 2000, 33, 4403–4410. [Google Scholar] [CrossRef]
- Veličković, J.; Ćoseva, S.; Fort, R.J. Solution properties of poly(dicyclohexyl itaconate). Eur. Polym. J. 1975, 11, 377–380. [Google Scholar] [CrossRef]
- Strazielle, C.; Benoit, H.; Vogl, O. Preparation et characterisation des polymers tete-a-tete—VI: Proprieties physicochimiques du polystyrene tete-a-tete en solution diluee. Comparaison avec des polystyrenes de structure differente. Eur. Polym. J. 1978, 14, 331–334. [Google Scholar] [CrossRef]
- Andrews, R.J.; Grulke, E.A. Glass transition temperatures of polymers. In Polymer Handbook, 4th ed.; Brandrup, J., Immergut, E.H., Grulke, E.A., Abe, A., Bloch, D.R., Eds.; Wiley: New York, NY, USA, 1999; Volume 1, pp. 181–308. [Google Scholar]
- Arrighi, V.; Holmes, P.F.; McEwen, I.J.; Qian, H.; Terrill, N.J. Order in amorphous di-n-alkyl itaconate polymers, copolymers, and blends. J. Polym. Sci. B Polym. Phys. 2004, 42, 4000–4016. [Google Scholar] [CrossRef]
- Cowie, J.M.G. Physical properties of polymers based on itaconic acid. Pure Appl. Chem. 1979, 51, 2331–2343. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample ID a | fDBI,0 a | T (°C) | [DBI] (M) | [S] (M) | [NHS-BB] (M) | [Dioxane] (M) |
|---|---|---|---|---|---|---|
| DBI/S-110-10 | 0.10 | 110 | 0.41 | 3.62 | 0.026 | 5.54 |
| DBI/S-110-20 | 0.20 | 110 | 0.73 | 2.91 | 0.023 | 5.58 |
| DBI/S-110-30 | 0.29 | 110 | 0.99 | 2.33 | 0.021 | 5.61 |
| DBI/S-110-40 | 0.40 | 110 | 1.22 | 1.83 | 0.019 | 5.64 |
| DBI/S-110-50 | 0.51 | 110 | 1.41 | 1.41 | 0.018 | 5.66 |
| DBI/S-110-60 | 0.60 | 110 | 1.58 | 1.05 | 0.017 | 5.68 |
| DBI/S-110-70 | 0.71 | 110 | 1.72 | 0.73 | 0.016 | 5.69 |
| DBI/S-110-80 | 0.80 | 110 | 1.84 | 0.46 | 0.015 | 5.71 |
| DB1/S-100-50 | 0.50 | 100 | 1.41 | 1.41 | 0.018 | 5.66 |
| DBI/S-80-50 | 0.50 | 80 | 1.41 | 1.41 | 0.018 | 5.66 |
| DBI/S-70-50 | 0.50 | 70 | 1.41 | 1.41 | 0.018 | 5.66 |
| DBI/S-70-60 | 0.60 | 70 | 1.58 | 1.05 | 0.017 | 5.68 |
| DBI/S-70-70 | 0.70 | 70 | 1.72 | 0.73 | 0.016 | 5.69 |
| DBI/S-70-80 | 0.80 | 70 | 1.84 | 0.46 | 0.015 | 5.71 |
| DBI/S-70-90 | 0.90 | 70 | 1.96 | 0.22 | 0.014 | 5.72 |
| Sample ID a | fDBI,0 | Time (min) | X b | FDBI c | Mn (kg mol−1) d | Đ d |
|---|---|---|---|---|---|---|
| DBI/S-110-10 | 0.10 | 120 | 0.51 | 0.13 | 11.2 | 1.28 |
| DBI/S-110-20 | 0.20 | 140 | 0.61 | 0.25 | 12.3 | 1.37 |
| DBI/S-110-30 | 0.29 | 170 | 0.80 | 0.30 | 9.9 | 1.57 |
| DBI/S-110-40 | 0.40 | 200 | 0.91 | 0.40 | 7.6 | 1.72 |
| DBI/S-110-50 | 0.51 | 180 | 0.87 | 0.45 | 5.5 | 1.68 |
| DBI/S-110-60 | 0.60 | 180 | 0.71 | 0.50 | 5.0 | 1.62 |
| DBI/S-110-70 | 0.71 | 230 | 0.75 | 0.60 | 3.7 | 1.52 |
| DBI/S-110-80 | 0.80 | 240 | 0.50 | 0.65 | 2.6 | 1.36 |
| Sample ID a | fDBI,0 | Time (min) | X b | Mn (kg mol−1) c | Đ c |
|---|---|---|---|---|---|
| DBI/S-110-50 | 0.51 | 180 | 0.87 | 5.5 | 1.68 |
| DBI/S-100-50 | 0.50 | 180 | 0.59 | 7.1 | 1.53 |
| DBI/S-80-50 | 0.50 | 340 | 0.41 | 5.8 | 1.42 |
| DBI/S-70-50 | 0.50 | 4320 | 0.47 | 8.0 | 1.42 |
| Sample ID a | fDBI,0 | Time (min) | X b | Mn (kg mol−1) c | Đ c |
|---|---|---|---|---|---|
| DBI/S-70-60 | 0.60 | 4320 | 0.56 | 6.9 | 1.63 |
| DBI/S-70-70 | 0.70 | 4320 | 0.58 | 7.2 | 1.41 |
| DBI/S-70-80 | 0.80 | 4320 | 0.66 | 5.9 | 1.33 |
| DBI/S-70-90 | 0.90 | 4320 | 0.38 | 3.2 | 1.38 |
| Experiment | Macroinitiator | Product | ||
|---|---|---|---|---|
| Mn (kg mol−1) | Đ | Mn (kg mol−1) | Đ | |
| DBI/S-110-20-b-S | 12.3 | 1.37 | 22.1 | 2.99 |
| DBI/S-110-80-b-S | 2.6 | 1.36 | 19.2 | 1.61 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kardan, S.; Garcia Valdez, O.; Métafiot, A.; Maric, M. Nitroxide-Mediated Copolymerization of Itaconate Esters with Styrene. Processes 2019, 7, 254. https://doi.org/10.3390/pr7050254
Kardan S, Garcia Valdez O, Métafiot A, Maric M. Nitroxide-Mediated Copolymerization of Itaconate Esters with Styrene. Processes. 2019; 7(5):254. https://doi.org/10.3390/pr7050254
Chicago/Turabian StyleKardan, Sepehr, Omar Garcia Valdez, Adrien Métafiot, and Milan Maric. 2019. "Nitroxide-Mediated Copolymerization of Itaconate Esters with Styrene" Processes 7, no. 5: 254. https://doi.org/10.3390/pr7050254