
Selective and Efficient Catalytic Oxygenation of Alkyl Aromatics Employing H2O2 Catalyzed by Simple Porphyrin Iron(II) under Mild Conditions
 and
and
Abstract
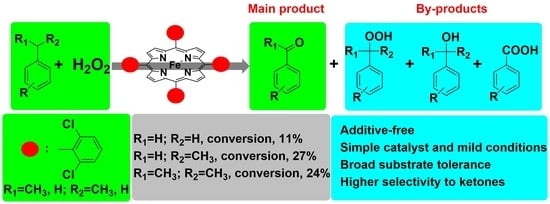
:
1. Introduction
2. Experimental Section
2.1. Syntheses of Metalloporphyrins
2.2. Autoxidation of Ethylbenzene
2.3. Catalytic Oxygenation of Ethylbenzene and Its Derivates
2.4. Kinetic Study
2.5. Products Analyses
3. Results and Discussion
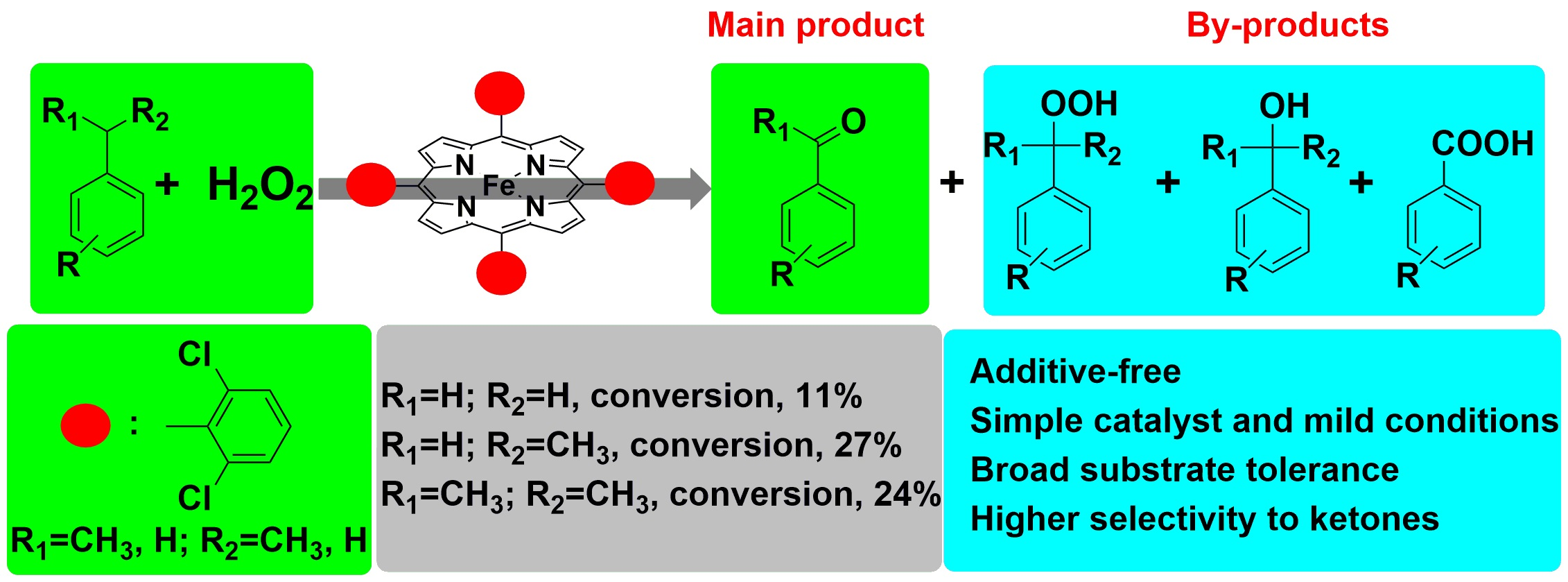
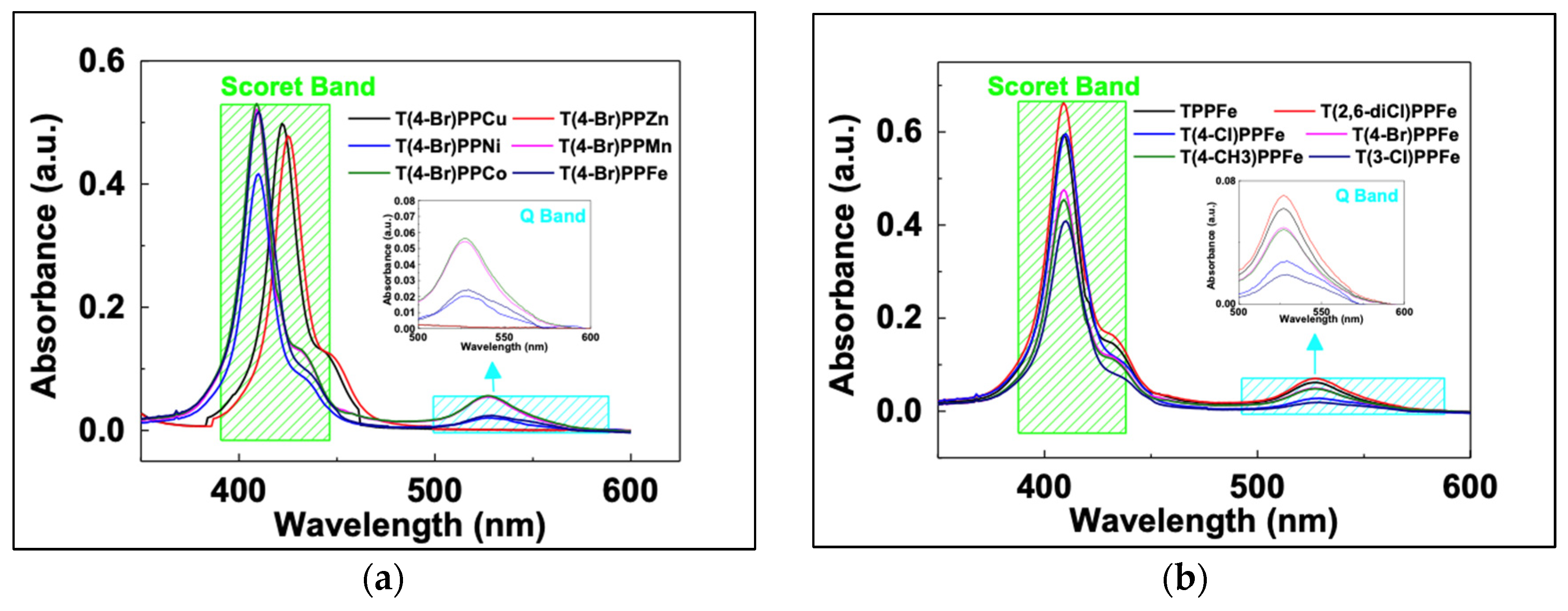
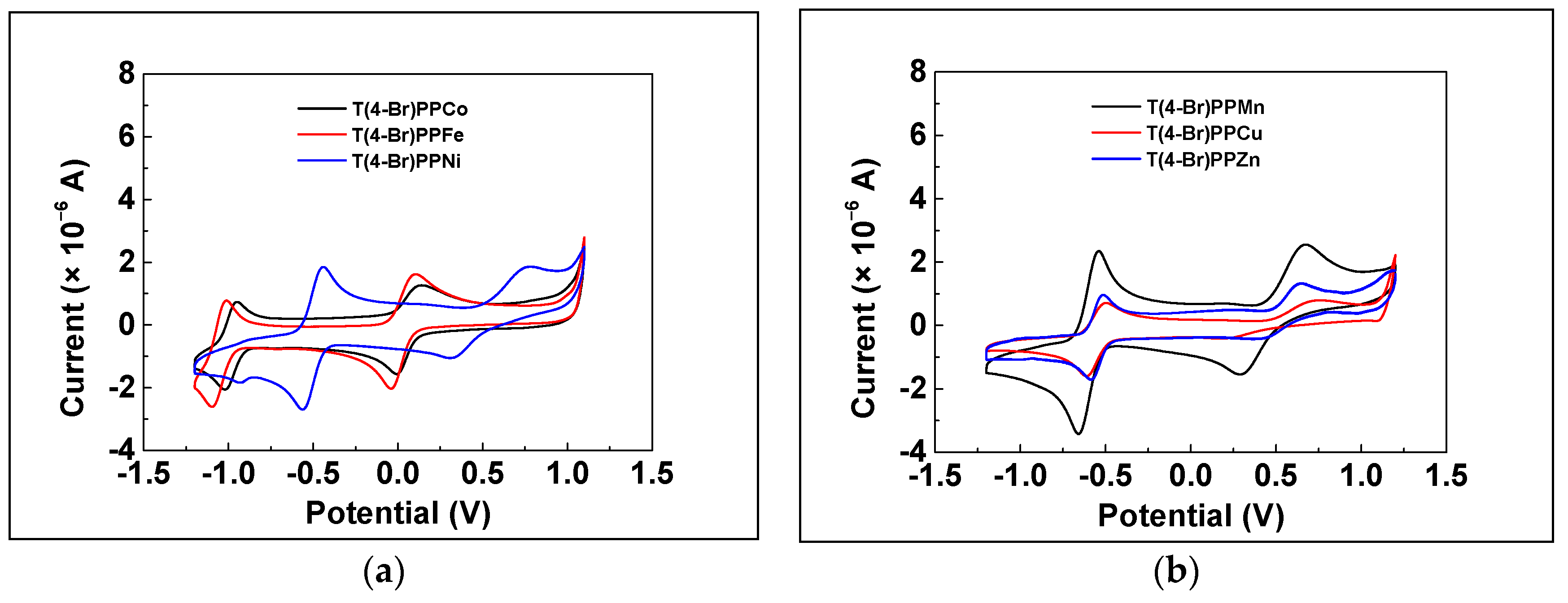
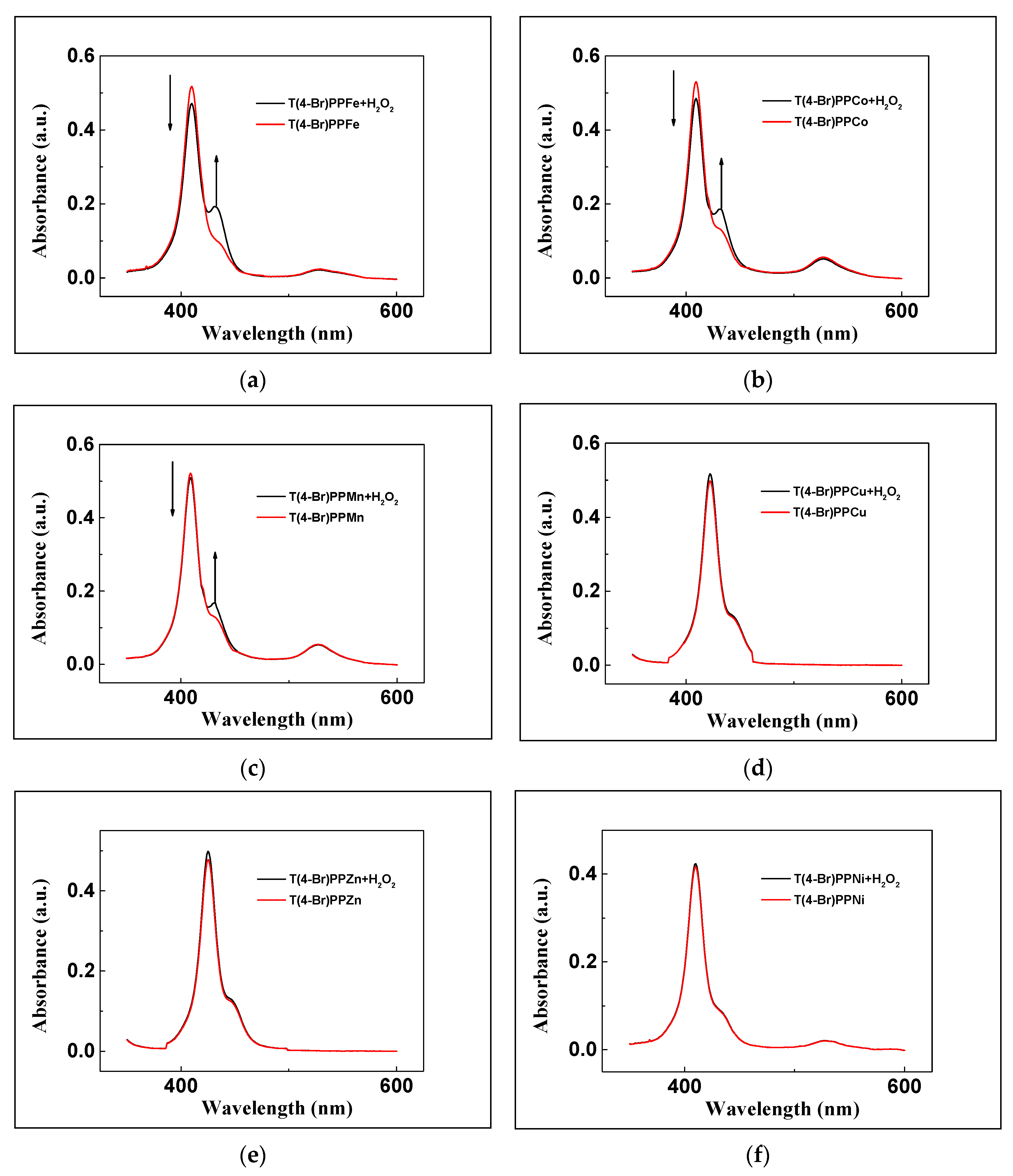
3.1. Characterizations
3.2. Preliminary Exploratory Research
3.3. Effect of Central Metal on Catalytic Oxygenation of Alkyl Aromatics
3.4. Effect of Porphyrin Ligands on Alkyl Aromatics Catalytic Oxygenation
3.5. Effect of Catalyst Loading and Oxidant Amount
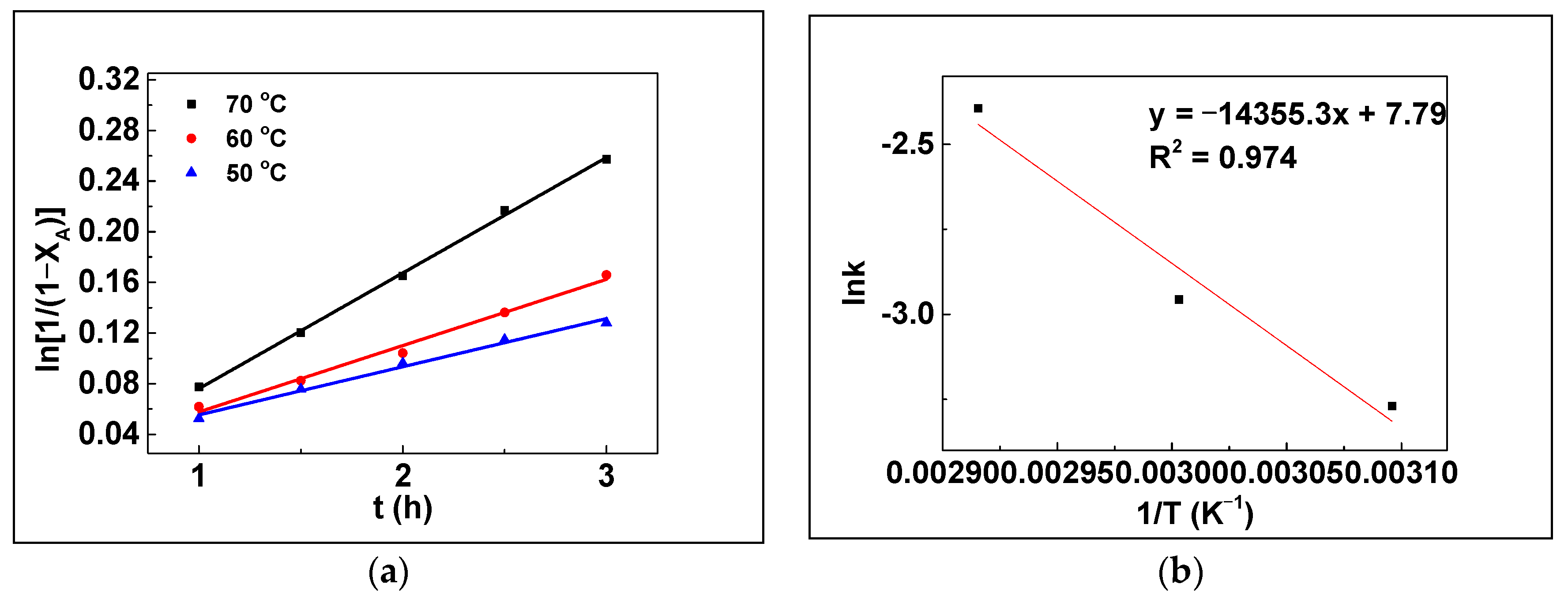
3.6. Kinetic Study
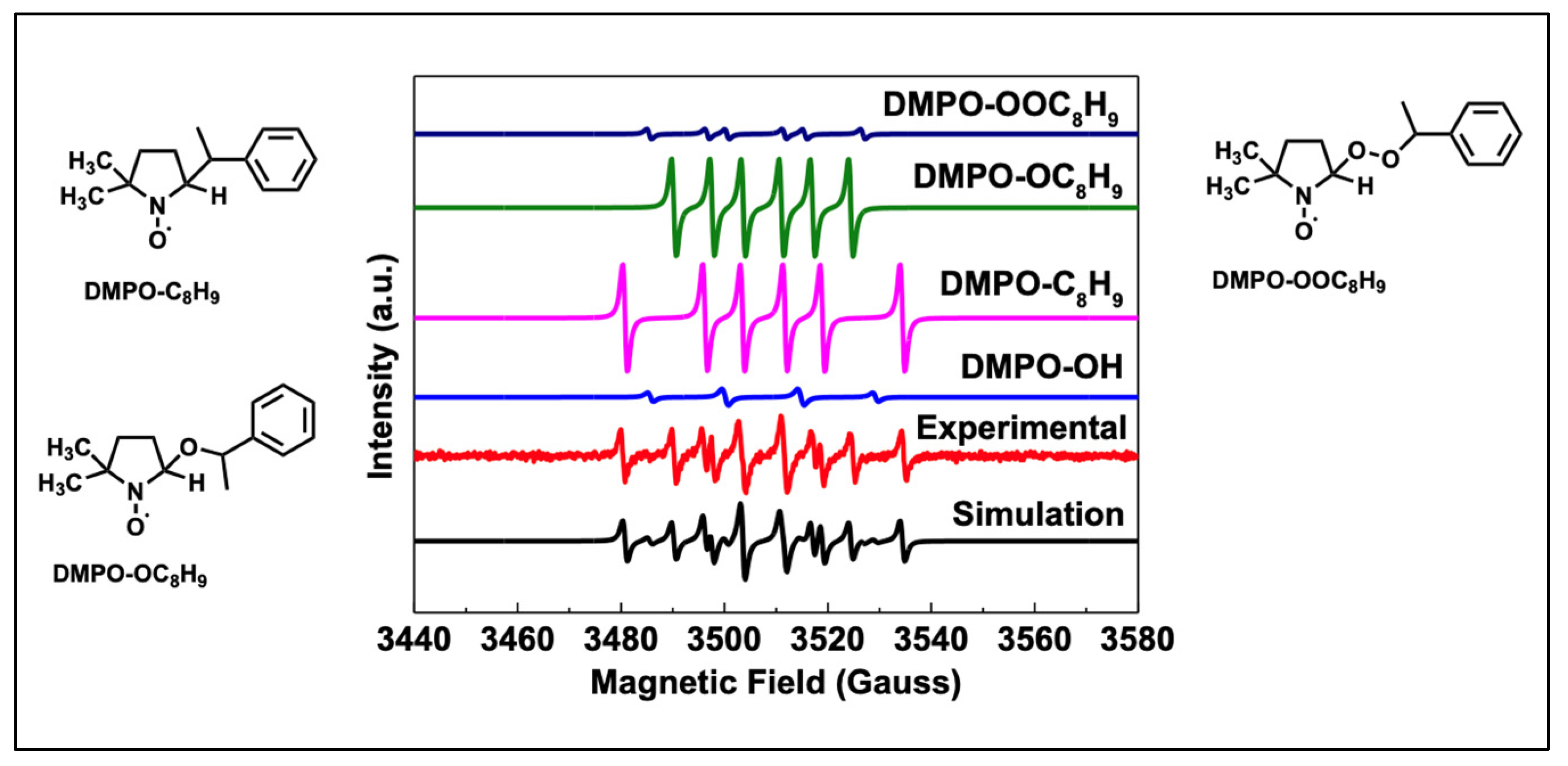
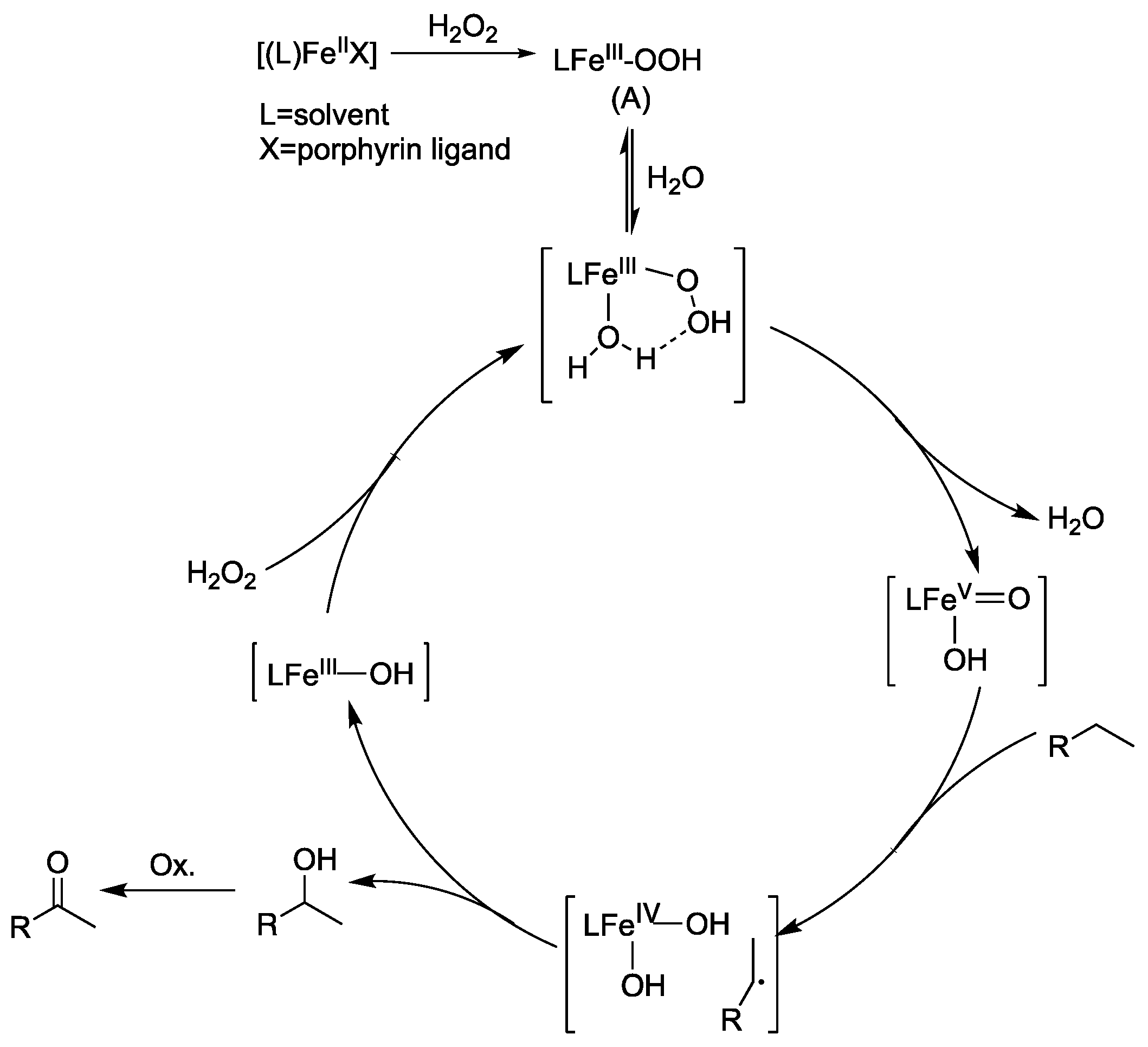
3.7. Mechanism on the Reaction Pathways
3.8. Comparison with Different H2O2 Catalytic Systems and Substrate Scope
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Abazid, A.H.; Clamor, N.; Nachtsheim, B.J. An Enantioconvergent Benzylic Hydroxylation Using a Chiral Aryl Iodide in a Dual Activation Mode. ACS Catal. 2020, 10, 8042–8048. [Google Scholar] [CrossRef]
- Wang, A.; Zhou, W.; Sun, Z.; Zhang, Z.; Zhang, Z.; He, M.; Chen, Q. Mn(III) active site in hydrotalcite efficiently catalyzes the oxidation of alkylarenes with molecular oxygen. Mol. Catal. 2021, 499, 111276. [Google Scholar] [CrossRef]
- Wang, H.X.; Wu, L.; Zheng, B.; Du, L.; To, W.P.; Ko, C.H.; Phillips, D.L.; Che, C.M. C—H Activation by an Iron-Nitrido Bis-Pocket Porphyrin Species. Angew. Chem. Int. Ed. 2021, 60, 4796–4803. [Google Scholar] [CrossRef] [PubMed]
- Bo, C.-B.; Bu, Q.; Li, X.; Ma, G.; Wei, D.; Guo, C.; Dai, B.; Liu, N. Highly Active and Robust Ruthenium Complexes Based on Hemilability of Hybrid Ligands for C—H Oxidation. J. Org. Chem. 2020, 85, 4324–4334. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Wang, Y.; Wang, X.; Wang, Y.; Liu, Y.; Huang, K.; Hu, J.; Duan, L.; Hu, C.; Liu, J. Selective Oxidation of Benzylic C—H Bonds Catalyzed by Cu(II)/{PMo12}. J. Org. Chem. 2020, 85, 3101–3109. [Google Scholar] [CrossRef]
- Lubov, D.P.; Lyakin, O.Y.; Samsonenko, D.G.; Rybalova, T.V.; Talsi, E.P.; Bryliakov, K.P. Palladium aminopyridine complexes catalyzed selective benzylic C—H oxidations with peracetic acid. Dalton Trans. 2020, 49, 11150–11156. [Google Scholar] [CrossRef]
- Mahmoudi, B.; Rostami, A.; Kazemnejadi, M.; Hamah-Ameen, B.A. Catalytic oxidation of alcohols and alkyl benzenes to carbonyls using Fe3O4@SiO2@(TEMPO)-co-(Chlorophyll-CoIII) as a bi-functional, self-co-oxidant nanocatalyst. Green Chem. 2020, 22, 6600–6613. [Google Scholar] [CrossRef]
- Xu, S.; Shi, G.; Feng, Y.; Chen, C.; Ji, L. Highly efficient transformation of ethylbenzene into acetophenone catalyzed by NHPI/Co(II) using molecular oxygen in hexafluoropropan-2-ol. Mol. Catal. 2020, 498, 111244. [Google Scholar] [CrossRef]
- Hu, X.; Sun, X.; Song, Q.; Zhu, Y.; Long, Y.; Dong, Z. N,S co-doped hierarchically porous carbon materials for efficient metal-free catalysis. Green Chem. 2020, 22, 742–752. [Google Scholar] [CrossRef]
- Wang, Z.; Jiang, Y.; Huo, H.; Hu, Y.; Xu, X.; Wang, P.; Yang, Y.; Lin, K. Synthesis of three-dimensional nitrogen doped meso/macroporous carbon beads for heterogeneous catalytic solvent-free oxidation of ethylbenzene. Carbon 2020, 158, 226–237. [Google Scholar] [CrossRef]
- Ottenbacher, R.V.; Talsi, E.P.; Bryliakov, K.P. Highly enantioselective undirected catalytic hydroxylation of benzylic CH2 groups with H2O2. J. Catal. 2020, 390, 170–177. [Google Scholar] [CrossRef]
- Ji, D.; Xi, N.; Li, G.; Dong, P.; Li, H.; Li, H.; Li, C.; Wang, P.; Zhao, Y. Hydrotalcite-based CoxNiyAl1Ox mixed oxide as a highly efficient catalyst for selective ethylbenzene oxidation. Mol. Catal. 2021, 508, 111579. [Google Scholar] [CrossRef]
- Selvaraj, M.; Bhaumik, A.; Assiri, M.A.; Subrahmanyam, C.; Ha, C.-S. Green oxidation of alkylaromatics using molecular oxygen over mesoporous manganese silicate catalysts. Dalton Trans. 2020, 49, 9710–9718. [Google Scholar] [CrossRef]
- Nie, R.; Chen, J.; Chen, M.; Qi, Z.; Goh, T.W.; Ma, T.; Zhou, L.; Pei, Y.; Huang, W. Aerobic oxidation of the C—H bond under ambient conditions using highly dispersed Co over highly porous N-doped carbon. Green Chem. 2019, 21, 1461–1466. [Google Scholar] [CrossRef]
- Wu, H.; Song, J.; Xie, C.; Hu, Y.; Liu, S.; Han, B. Preparation of Copper Phosphate from Naturally Occurring Phytic Acid as an Advanced Catalyst for Oxidation of Aromatic Benzyl Compounds. ACS Sustain. Chem. Eng. 2018, 6, 13670–13675. [Google Scholar] [CrossRef]
- Trehoux, A.; Guillot, R.; Clemancey, M.; Blondin, G.; Latour, J.-M.; Mahy, J.-P.; Avenier, F. Bioinspired symmetrical and unsymmetrical diiron complexes for selective oxidation catalysis with hydrogen peroxide. Dalton Trans. 2020, 49, 16657–16661. [Google Scholar] [CrossRef]
- Shen, H.-M.; Liu, L.; Qi, B.; Hu, M.-Y.; Ye, H.-L.; She, Y.-B. Efficient and selective oxidation of secondary benzylic C—H bonds to ketones with O2 catalyzed by metalloporphyrins under solvent-free and additive-free conditions. Mol. Catal. 2020, 493, 111102. [Google Scholar] [CrossRef]
- Shen, H.-M.; Hu, M.-Y.; Qi, B.; Liu, L.; Ye, H.-L.; She, Y.-B. Efficient and selective oxidation of tertiary benzylic C—H bonds with O2 catalyzed by metalloporphyrins under mild and solvent-free conditions. Appl. Catal. A Gen. 2020, 599, 117599. [Google Scholar] [CrossRef]
- Feng, D.; Gu, Z.Y.; Li, J.R.; Jiang, H.L.; Wei, Z.; Zhou, H.C. Zirconium-metalloporphyrin PCN-222: Mesoporous metal-organic frameworks with ultrahigh stability as biomimetic catalysts. Angew. Chem. Int. Ed. 2012, 51, 10453–10456. [Google Scholar] [CrossRef]
- Jiang, X.; Gou, F.; Jing, H. Alternating copolymerization of CO2 and propylene oxide catalyzed by C-2v-porphyrin cobalt: Selectivity control and a kinetic study. J. Catal. 2014, 313, 159–167. [Google Scholar] [CrossRef]
- Sheng, N.; Gu, B.; Ren, B.; Zhang, J.; Wang, Y.; Wang, J.; Sha, J. A series of polycyclic aromatic hydrocarbon-substituted metal-free porphyrins: Substituent effect on two-photon absorption property. Dye Pigment 2017, 142, 116–120. [Google Scholar] [CrossRef]
- Wang, Z.; Zhang, J.-H.; Jiang, J.-J.; Wang, H.-P.; Wei, Z.-W.; Zhu, X.; Pan, M.; Su, C.-Y. A stable metal cluster-metalloporphyrin MOF with high capacity for cationic dye removal. J. Mater. Chem. A 2018, 6, 17698–17705. [Google Scholar] [CrossRef]
- Bichan, N.; Ovchenkova, E.; Ksenofontov, A.; Kudryakova, N.; Semeikin, A.; Lomova, T. Self-organizing donor-acceptor assemblies of cobalt(II) porphyrin ligated with gold(III) porphyrin or fullero[60]pyrrolidine in liquid medium. J. Mol. Liq. 2021, 326, 115306. [Google Scholar] [CrossRef]
- Chen, W.-T.; Zhang, Z.-X.; Lin, L.-Z.; Sui, Y.; Liu, D.-S.; Chen, H.-L. Preparation, Crystal Structures, and Properties of a Series of Crystalline Tetra(4-sulfonatophenyl)porphyrinato Histidine 4f-3d Porphyrinic Compounds. Cryst. Growth Des. 2018, 18, 5456–5464. [Google Scholar] [CrossRef]
- Deng, W.; Luo, W.-P.; Tan, Z.; Liu, Q.; Liu, Z.-M.; Guo, C.-C. Remarkable effect of simple aliphatic alcohols on the controlled aerobic oxidation of toluene catalyzed by (T(p-Cl)PP)MnF/NHPI. J. Mol. Catal. A Chem. 2013, 372, 84–89. [Google Scholar] [CrossRef]
- Mondal, B.; Saha, S.; Borah, D.; Mazumdar, R.; Mondal, B. Nitric Oxide Dioxygenase Activity of a Nitrosyl Complex of Cobalt(II) Porphyrinate in the Presence of Hydrogen Peroxide via Putative Peroxynitrite Intermediate. Inorg. Chem. 2019, 58, 1234–1240. [Google Scholar] [CrossRef] [PubMed]
- Ottenbacher, R.V.; Talsi, E.P.; Bryliakov, K.P. Mechanism of Selective C—H Hydroxylation Mediated by Manganese Aminopyridine Enzyme Models. ACS Catal. 2014, 5, 39–44. [Google Scholar] [CrossRef]
- Calvete, M.J.F.; Piñeiro, M.; Dias, L.D.; Pereira, M.M. Hydrogen Peroxide and Metalloporphyrins in Oxidation Catalysis: Old Dogs with Some New Tricks. Chemcatchem 2018, 10, 3615–3635. [Google Scholar] [CrossRef]
- Tkachenko, N.V.; Ottenbacher, R.V.; Lyakin, O.Y.; Zima, A.M.; Samsonenko, D.G.; Talsi, E.P.; Bryliakov, K.P. Highly Efficient Aromatic C—H Oxidation with H2O2 in the Presence of Iron Complexes of the PDP Family. ChemCatChem 2018, 10, 4052–4057. [Google Scholar] [CrossRef]
- Yu, H.; Zhao, Q.; Wei, Z.; Wu, Z.; Li, Q.; Han, S.; Wei, Y. Iron-catalyzed oxidative functionalization of C(sp3)-H bonds under bromide-synergized mild conditions. Chem. Commun. 2019, 55, 7840–7843. [Google Scholar] [CrossRef]
- Tan, J.; Liu, X.B.; Chen, W.F.; Hu, Y.L. Synthesis of Magnetically Separable Nanocatalyst CoFe2O4@SiO2@MIL-53(Fe) for Highly Efficient and Selective Oxidation of Alcohols and Benzylic Compounds with Hydrogen Peroxide. Chemistryselect 2019, 4, 8477–8481. [Google Scholar] [CrossRef]
- Arafa, W.A.A. Sonochemical Preparation of Dipicolinamide Mn-complexes and Their Application as Catalysts Towards Sono-synthesis of Ketones. J. Heterocycl. Chem. 2019, 56, 1403–1412. [Google Scholar] [CrossRef]
- Wu, C.; Liu, B.; Geng, X.; Zhang, Z.; Liu, S.; Hu, Q. Selective catalytic oxidation of aromatic substrates employing mononuclear copper(II) catalyst with H2O2. Polyhedron 2019, 158, 334–341. [Google Scholar] [CrossRef]
- Chakraborty, T.; Chakraborty, A.; Maity, S.; Das, D.; Chattopadhyay, T. Conglomerated system of Ag nanoparticles decorated Al2O3 supported cobalt and copper complexes with enhanced catalytic activity for oxidation reactions. Mol. Catal. 2019, 462, 104–113. [Google Scholar] [CrossRef]
- Maurya, M.R.; Sarkar, B.; Kumar, A.; Ribeiro, N.; Miliute, A.; Pessoa, J.C. New thiosemicarbazide and dithiocarbazate based oxidovanadium(IV) and dioxidovanadium(V) complexes. Reactivity and catalytic potential. N. J. Chem. 2019, 43, 17620–17635. [Google Scholar] [CrossRef]
- Dey, D.; Patra, M.; Al-Hunaiti, A.; Yadav, H.R.; Al-Mherat, A.; Arar, S.; Maji, M.; Choudhury, A.R.; Biswas, B. Synthesis, structural characterization and C—H activation property of a tetra-iron(III) cluster. J. Mol. Struct. 2019, 1180, 220–226. [Google Scholar] [CrossRef]
- Hosseinzadeh, R.; Mavvaji, M.; Tajbakhsh, M.; Lasemi, Z.; Aghili, N. Selective Oxidation of Hydrocarbons and Alcohols Using Phen-MCM-41 as an Efficient Co-Catalyst in Combination with NHPI-Based Nano-Magnetic Catalyst. Org. Prep. Proced. Int. 2020, 52, 99–109. [Google Scholar] [CrossRef]
- Keshipour, S.; Al-Azmi, A. Synthesis and catalytic application of Pd/PdO/Fe3O4@polymer-like graphene quantum dots. Appl. Organomet. Chem. 2019, 34, 5311–5319. [Google Scholar]
- Al-Hunaiti, A.; Al-Said, N.; Halawani, L.; Abu Haija, M.; Baqaien, R.; Taher, D. Synthesis of magnetic CuFe2O4 nanoparticles as green catalyst for toluene oxidation under solvent-free conditions. Arab. J. Chem. 2020, 13, 4945–4953. [Google Scholar] [CrossRef]
- Ticconi, B.; Capocasa, G.; Cerrato, A.; Di Stefano, S.; Lapi, A.; Marincioni, B.; Olivo, G.; Lanzalunga, O. Insight into the chemoselective aromatic vs. side-chain hydroxylation of alkylaromatics with H2O2 catalyzed by a non-heme imine-based iron complex. Catal. Sci. Technol. 2021, 11, 171–178. [Google Scholar] [CrossRef]
- Verma, S.; Nasir Baig, R.B.; Nadagouda, M.N.; Varma, R.S. Photocatalytic C—H Activation of Hydrocarbons over VO@g-C3N4. ACS Sustain. Chem. Eng. 2016, 4, 2333–2336. [Google Scholar] [CrossRef]
- Nouri, S.H.; Hosseini-Monfared, H. Highly efficient and green oxidation of alkanes and alkylaromatics with hydrogen peroxide catalysed by silver and vanadyl on mesoporous silica-coated magnetite. Appl. Organomet. Chem. 2017, 31, 3649–3655. [Google Scholar] [CrossRef]
- Wang, W.; Xu, D.; Sun, Q.; Sun, W. Efficient Aliphatic C−H Bond Oxidation Catalyzed by Manganese Complexes with Hydrogen Peroxide. Chem. Asian J. 2018, 13, 2458–2464. [Google Scholar] [CrossRef] [PubMed]
- Patel, P.; Nandi, S.; Menapara, T.; Biradar, A.V.; Nagarale, R.K.; Khan, N.H.; Kureshy, R.I. Glycoluril: A heterogeneous organocatalyst for oxidation of alcohols and benzylic sp3 carbons. Appl. Catal. A Gen. 2018, 565, 127–134. [Google Scholar] [CrossRef]
- Basu, P.; Dey, T.K.; Ghosh, A.; Islam, S.M. Designing of a New Heterogeneous Polymer Supported Naphthyl-Azo Iron Catalyst for the Selective Oxidation of Substituted Methyl Benzenes. J. Inorg. Organomet. Polym. Mater. 2018, 28, 1158–1170. [Google Scholar] [CrossRef]
- Su, E.; Guven, A.; Kani, I. Oxygen bridged Homobinuclear Mn(II) compounds with Anthranilic acid: Theoretical calculations, oxidation and catalase activity. Appl. Organomet. Chem. 2017, 32, 4105–4122. [Google Scholar] [CrossRef]
- Tkachenko, N.V.; Lyakin, O.Y.; Zima, A.M.; Talsi, E.P.; Bryliakov, K.P. Effect of different carboxylic acids on the aromatic hydroxylation with H2O2 in the presence of an iron aminopyridine complex. J. Organomet. Chem. 2018, 871, 130–134. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | |||||||
|---|---|---|---|---|---|---|---|
| Entry | Catalysts | Temperature (°C) | Conversion (%) | Selectivity (%) | |||
| R1=O | R1-OH | R1-OOH | R2-COOH | ||||
| 1 | - | 50 | <1% | - | - | - | - |
| 2 | - | 55 | <1% | - | - | - | - |
| 3 | - | 60 | <1% | - | - | - | - |
| 4 | - | 65 | <1% | - | - | - | - |
| 5 | - | 70 | <1% | - | - | - | - |
| 6 | TPPFe | 50 | 12.50 | 74 | 26 | - | - |
| 7 | TPPFe | 55 | 15.35 | 76 | 24 | - | - |
| 8 | TPPFe | 60 | 17.15 | 77 | 23 | - | - |
| 9 | TPPFe | 65 | 19.46 | 78 | 22 | - | - |
| 10 | TPPFe | 70 | 19.71 | 79 | 21 | - | - |
| 11 | T(4-Br)PPFe | 50 | 9.26 | 77 | 23 | - | - |
| 12 | T(4-Br)PPFe | 55 | 12.08 | 80 | 20 | - | - |
| 13 | T(4-Br)PPFe | 60 | 12.83 | 83 | 17 | - | - |
| 14 | T(4-Br)PPFe | 65 | 13.66 | 87 | 13 | - | - |
| 15 | T(4-Br)PPFe | 70 | 14.35 | 93 | 7 | - | - |
| 16 | TPPFe b | 70 | 19.52 | 79 | 21 | - | - |
| 17 | T(4-Br)PPFe b | 70 | 14.25 | 92 | 8 | - | - |
| 18 | TPP | 70 | <1% | - | - | - | - |
| 19 | T(4-Br)PP | 70 | <1% | - | - | - | - |
| 20 | Fe(OAc)2 | 70 | <1% | - | - | - | - |
 | ||||||
|---|---|---|---|---|---|---|
| Entry | Catalysts | Conversion (%) | Selectivity (%) | |||
| R1=O | R1-OH | R1-OOH | R2-COOH | |||
| 1 | TPPFe | 19.71 | 79 | 21 | - | - |
| 2 | TPPCo | <1% | - | - | - | - |
| 3 | TPPMn | <1% | - | - | - | - |
| 4 | TPPNi | <1% | - | - | - | - |
| 5 | TPPCu | <1% | - | - | - | - |
| 6 | TPPZn | <1% | - | - | - | - |
| 7 | T(4-Br)PPFe | 14.35 | 93 | 7 | - | - |
| 8 | T(4-Br)PPCo | <1% | - | - | - | - |
| 9 | T(4-Br)PPMn | <1% | - | - | - | - |
| 10 | T(4-Br)PPNi | <1% | - | - | - | - |
| 11 | T(4-Br)PPCu | <1% | - | - | - | - |
| 12 | T(4-Br)PPZn | <1% | - | - | - | - |
 | ||||||
|---|---|---|---|---|---|---|
| Entry | Catalysts | Conversion (%) | Selectivity (%) | |||
| R1=O | R1-OH | R1-OOH | R2-COOH | |||
| 1 | TPPFe | 19.71 | 79 | 21 | - | - |
| 2 | T(2-Cl)PPFe | <1% | - | - | - | - |
| 3 | T(3-Cl)PPFe | 17.46 | 86 | 14 | - | - |
| 4 | T(4-Cl)PPFe | 20.84 | 82 | 18 | - | - |
| 5 | T(2,6-diCl)PPFe | 27.44 | 85 | 15 | - | - |
| 6 | T(2-CH3)PPFe | <1% | - | - | - | - |
| 7 | T(3-CH3)PPFe | 13.87 | 82 | 18 | - | - |
| 8 | T(4-CH3)PPFe | 20.52 | 78 | 22 | - | - |
| 9 | T(3-OCH3)PPFe | 12.89 | 83 | 17 | - | - |
| 10 | T(4-OCH3)PPFe | <1% | - | - | - | - |
| 11 | T(3-F-4-Br)PPFe | 13.63 | 87 | 13 | - | - |
| 12 | T(2-F-4-Br)PPFe | 12.57 | 85 | 15 | - | - |
| 13 | T(2-Cl-4-Br)PPFe | 14.12 | 87 | 13 | - | - |
| 14 | T(3-Cl-4-Br)PPFe | 14.21 | 86 | 14 | - | - |
| 15 | T(2,3,6-triCl)PPFe | 3.87 | 85 | 15 | - | - |
| 16 | T(2,3,5-triCl)PPFe | 3.76 | 82 | 18 | - | - |
| 17 | T(2,3,6-triF)PPFe | 1.62 | 82 | 18 | - | - |
| 18 | T(2,3,5-triF)PPFe | 2.01 | 83 | 17 | - | - |
 | |||||||
|---|---|---|---|---|---|---|---|
| Entry | Catalysts | Catalyst Amount | Conversion (%) | Selectivity (%) | |||
| (%, mol/mol) | R1=O | R1-OH | R1-OOH | R2-COOH | |||
| 1 | TPPFe | 0.005 | 18.95 | 80 | 20 | - | - |
| 2 | TPPFe | 0.010 | 19.71 | 79 | 21 | - | - |
| 3 | TPPFe | 0.015 | 20.53 | 81 | 19 | - | - |
| 4 | TPPFe | 0.020 | 20.72 | 81 | 19 | - | - |
| 5 | TPPFe | 0.025 | 21.09 | 80 | 20 | - | - |
| 6 | T(2,6-diCl)PPFe | 0.005 | 26.70 | 85 | 15 | - | - |
| 7 | T(2,6-diCl)PPFe | 0.010 | 27.44 | 85 | 15 | - | - |
| 8 | T(2,6-diCl)PPFe | 0.015 | 27.61 | 83 | 17 | - | - |
| 9 | T(2,6-diCl)PPFe | 0.020 | 27.83 | 84 | 16 | - | - |
| 10 | T(2,6-diCl)PPFe | 0.025 | 27.94 | 86 | 14 | - | - |
 | |||||||
|---|---|---|---|---|---|---|---|
| Entry | Catalysts | Mole Ratio | Conversion (%) | Selectivity (%) | |||
| (H2O2: Substrate) | R1=O | R1-OH | R1-OOH | R2-COOH | |||
| 1 | TPPFe | 4 | 7.84 | 77 | 23 | - | - |
| 2 | TPPFe | 6 | 13.41 | 79 | 21 | - | - |
| 3 | TPPFe | 8 | 19.71 | 79 | 21 | - | - |
| 4 | TPPFe | 10 | 20.75 | 75 | 19 | - | 6 |
| 5 | TPPFe | 12 | 21.22 | 72 | 18 | - | 10 |
| 6 | TPPFe | 14 | 21.50 | 71 | 18 | - | 11 |
| 7 | T(2,6-diCl)PPFe | 4 | 10.11 | 85 | 15 | - | - |
| 8 | T(2,6-diCl)PPFe | 6 | 20.93 | 84 | 16 | - | - |
| 9 | T(2,6-diCl)PPFe | 8 | 27.44 | 85 | 15 | - | - |
| 10 | T(2,6-diCl)PPFe | 10 | 28.48 | 81 | 15 | - | 4 |
| 11 | T(2,6-diCl)PPFe | 12 | 28.99 | 78 | 14 | - | 8 |
| 12 | T(2,6-diCl)PPFe | 14 | 29.36 | 77 | 14 | - | 9 |
| Entry | Catalysts | Temp. (°C) | k (L⋅mol−1⋅h−1) | R2 | Average Intercepts | Ea (kJ/mol) |
|---|---|---|---|---|---|---|
| 1 | TPPFe | 50 | 0.0217 | 0.9920 | 0.0060 | 51.37 |
| 2 | 60 | 0.0306 | 0.9941 | |||
| 3 | 70 | 0.0665 | 0.9916 | |||
| 4 | T(4-Br)PPFe | 50 | 0.0117 | 0.9929 | −0.0044 | 53.31 |
| 5 | 60 | 0.0170 | 0.9936 | |||
| 6 | 70 | 0.0374 | 0.9964 | |||
| 7 | T(2,6-diCl)PPFe | 50 | 0.0380 | 0.9906 | 0.0025 | 40.24 |
| 8 | 60 | 0.0524 | 0.9906 | |||
| 9 | 70 | 0.0913 | 0.9988 |
 | ||||||
|---|---|---|---|---|---|---|
| Entry | Substrates | Conversion (%) | Selectivity (%) | |||
| R1=O | R1-OH | R1-OOH | R2-COOH | |||
| 1 |  | 27.44 | 85 | 15 | - | - |
| 2 |  | 14.73 | 50 | 50 | - | - |
| 3 |  | 26.71 | 81 | 19 | - | - |
| 4 |  | 25.28 | 83 | 17 | - | - |
| 5 |  | 50.49 | 90 | 10 | - | - |
| 6 |  | 65.67 | 76 | 24 | - | - |
| 7 |  | 73.78 | 90 | 10 | - | - |
| 8 |  | 36.30 | 88 | 12 | - | - |
| 9 |  | 38.26 | 84 | 16 | - | - |
| 10 |  | 40.65 | 80 | 20 | - | - |
| Entry | Main Products | Conditions | Conversion (%) | Selectivity (%) | Ref. |
|---|---|---|---|---|---|
| 1 |  | supported iron (0.3%, m/m), H2O2 (3.5 equiv), n-Bu4NBr, 1,4-dioxane, 70 °C, 24.0 h | 98 | 91 | [30] |
| 2 |  | nanocomposite CoFe2O4@SiO2@MIL-53(Fe) (1.2%, m/m), H2O2 (2.3 equiv), H2O, 25 °C, 4.0 h | 94 | 99 | [31] |
| 3 |  | Mn(II) complex (0.1%, mol/mol), H2O2 (5 equiv), H2O, AcOH (10 equiv), 70 °C, 10.0 h | 89 | 91 | [32] |
| 4 |  | Cu(II) complex (0.1%, m/m), H2O2 (1 equiv), CH3CN, 70 °C, 8.0 h | 47 | 98.0 | [33] |
| 5 |  | copper nanoparticle in Al2O3 supported Co(II) and Cu(II) complex (10%, m/m), H2O2 (1.25 equiv), CH3CN, 50 °C, 4.0 h | 95 | 95.0 | [34] |
| 6 |  | V(IV) and V(V) complex (1%, m/m), H2O2 (2.0 equiv), CH3CN, 80 °C, 20.0 h | 65 | 82 | [35] |
| 7 |  | Tetra-Fe (III) cluster (0.04%, m/m), H2O2 (2.5 equiv), AcOH (0.5 equiv), 32 °C, 3.0 h | 60 | 76 | [36] |
| 8 |  | Phen-MCM-41 (Co-Catalyst) (2.5%, m/m), H2O2 (2.0 equiv), NHPI, CH3CN, 80 °C, 4.5 h | 78 | 76 | [37] |
| 9 |  | Pd/PdO/Fe3O4@PGQD (0.4%, m/m), H2O2 (2.0 equiv), MeOH, 25 °C, 20.0 h | 77 | 99 | [38] |
| 10 |  | CuFe2O4 nanoparticles (0.02%, m/m), H2O2 (3.0 equiv), CH3CN, 60 °C, 24.0 h | 56 | 89 | [39] |
| 11 |  | Fe(II) complex (0.1%, m/m), H2O2 (0.2 equiv), CH3CN, 25 °C, 1.5 h | 80 | 56 | [40] |
| 12 |  | VO@g-C3N4 (10.0%, m/m), H2O2 (1.5 equiv), CH3CN, 25 °C, 12.0 h | 99 | 99 | [41] |
| 13 |  | nanospheres of magnetite (Fe3O4@m-SiO2) support FeSi/Ag/VO nanocomposite (5.0%, m/m), H2O2 (1.5 equiv), CH3CN, 60 °C, 8.0 h | 46 | 72 | [42] |
| 14 |  | Mn(II) complex (0.5%, m/m), H2O2 (3.5 equiv), AcOH (14 equiv), 0 °C, 0.5 h | 72 | 93 | [43] |
| 15 |  | heterogeneous organocatalyst glycoluril (5.0%, m/m), H2O2 (1.2 equiv), H2O, 60 °C, 3.0 h | 99 | 98 | [44] |
| 16 |  | iron-anchored naphthyl-azo catalyst (PS-Fe-NAPA) (1.0%, m/m), H2O2 (2.5 equiv), CH3CN, 60 °C, 7.0 h | 96 | 93 | [45] |
| 17 |  | oxygen bridged homobinuclear Mn(II) compounds (1.0%, m/m), H2O2 (2.5 equiv), CH3CN, 60 °C, 7.0 h | 79 | 99 | [46] |
| 18 |  | iron based catalyst [(PDP)Fe(OTf)2] (1.0%, m/m), H2O2 (4 equiv), EHA (10 equiv), 0 °C, 2.5 h | 19 | 95 | [47] |
| 19 |  | metalloporphyrins T(2,6-diCl)PPFe (10.0%, m/m), H2O2 (8.0 equiv), CH3CN, 70 °C, 12.0 h | 27 | 85 | This work |
| 20 |  | metalloporphyrins T(2,6-diCl)PPFe (10.0%, m/m), H2O2 (8.0 equiv), CH3CN, 70 °C, 12.0 h | 36 | 88 | This work |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, X.-Y.; He, B.; Zhang, Y.; Ni, J.-Y.; Liu, Q.-P.; Wang, M.; Shen, H.-M.; She, Y.-B. Selective and Efficient Catalytic Oxygenation of Alkyl Aromatics Employing H2O2 Catalyzed by Simple Porphyrin Iron(II) under Mild Conditions. Processes 2023, 11, 1187. https://doi.org/10.3390/pr11041187
Zhou X-Y, He B, Zhang Y, Ni J-Y, Liu Q-P, Wang M, Shen H-M, She Y-B. Selective and Efficient Catalytic Oxygenation of Alkyl Aromatics Employing H2O2 Catalyzed by Simple Porphyrin Iron(II) under Mild Conditions. Processes. 2023; 11(4):1187. https://doi.org/10.3390/pr11041187
Chicago/Turabian StyleZhou, Xin-Yan, Bin He, Yu Zhang, Jia-Ye Ni, Qiu-Ping Liu, Mei Wang, Hai-Min Shen, and Yuan-Bin She. 2023. "Selective and Efficient Catalytic Oxygenation of Alkyl Aromatics Employing H2O2 Catalyzed by Simple Porphyrin Iron(II) under Mild Conditions" Processes 11, no. 4: 1187. https://doi.org/10.3390/pr11041187