
Density Functional Theory Study on the Adsorption of Fe(OH)2+ on Kaolinite Surface in Water Environment
 ,
,
Abstract
:1. Introduction
2. Methods
3. Results and Discussion
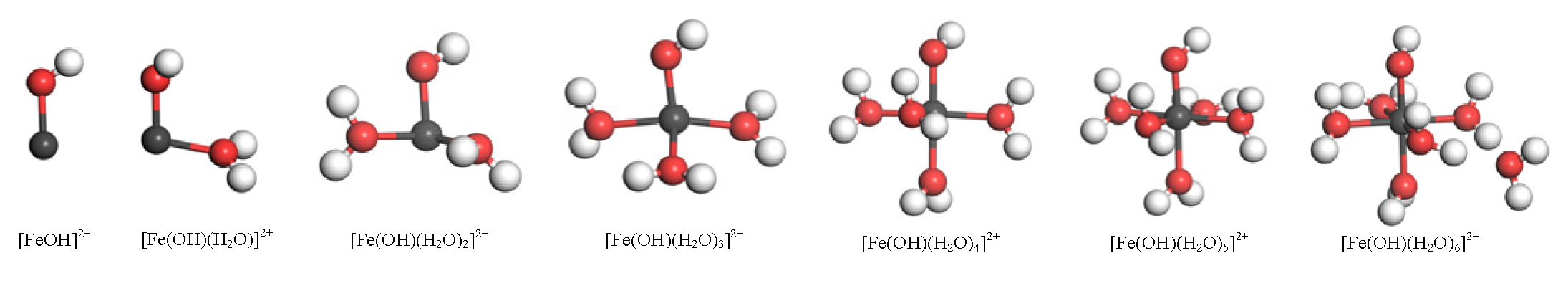
3.1. Hydration Structure and Properties of Fe(OH)2+
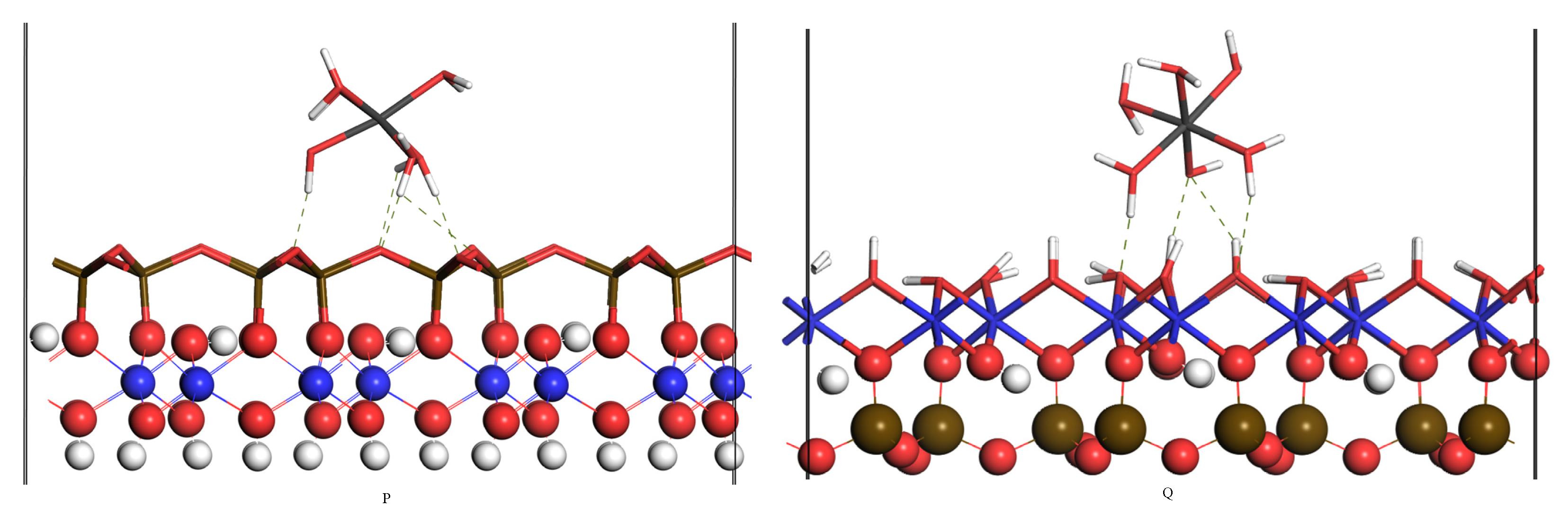
3.2. Outer Layer Adsorption Structure and Analysis
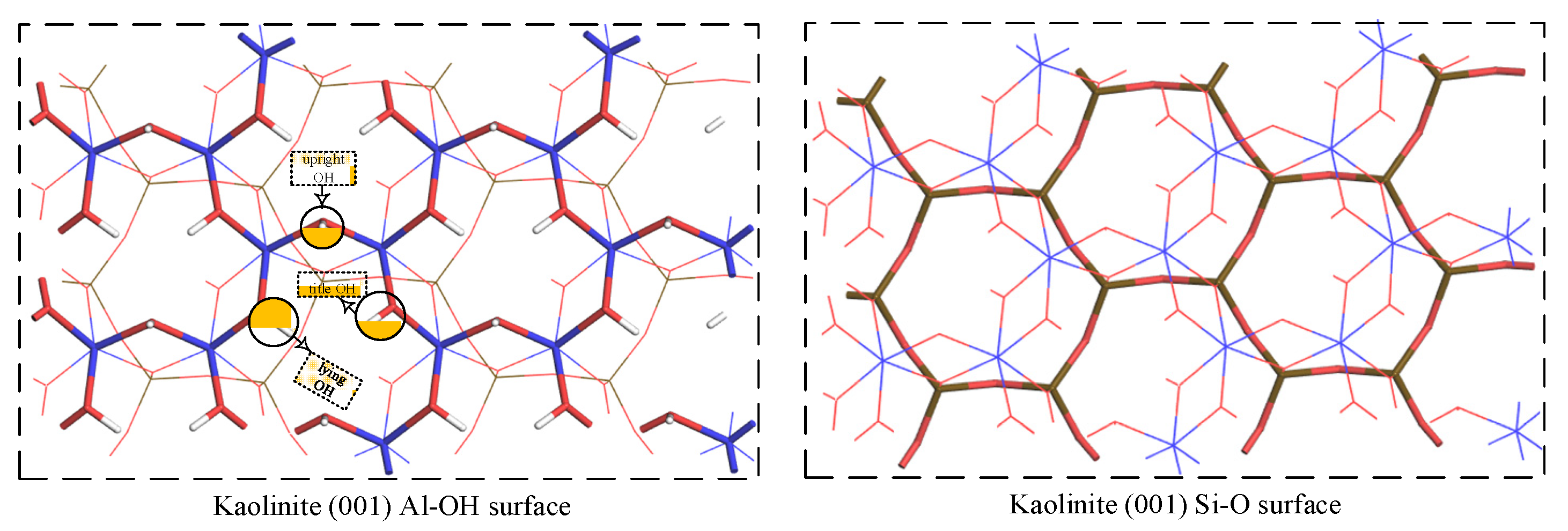
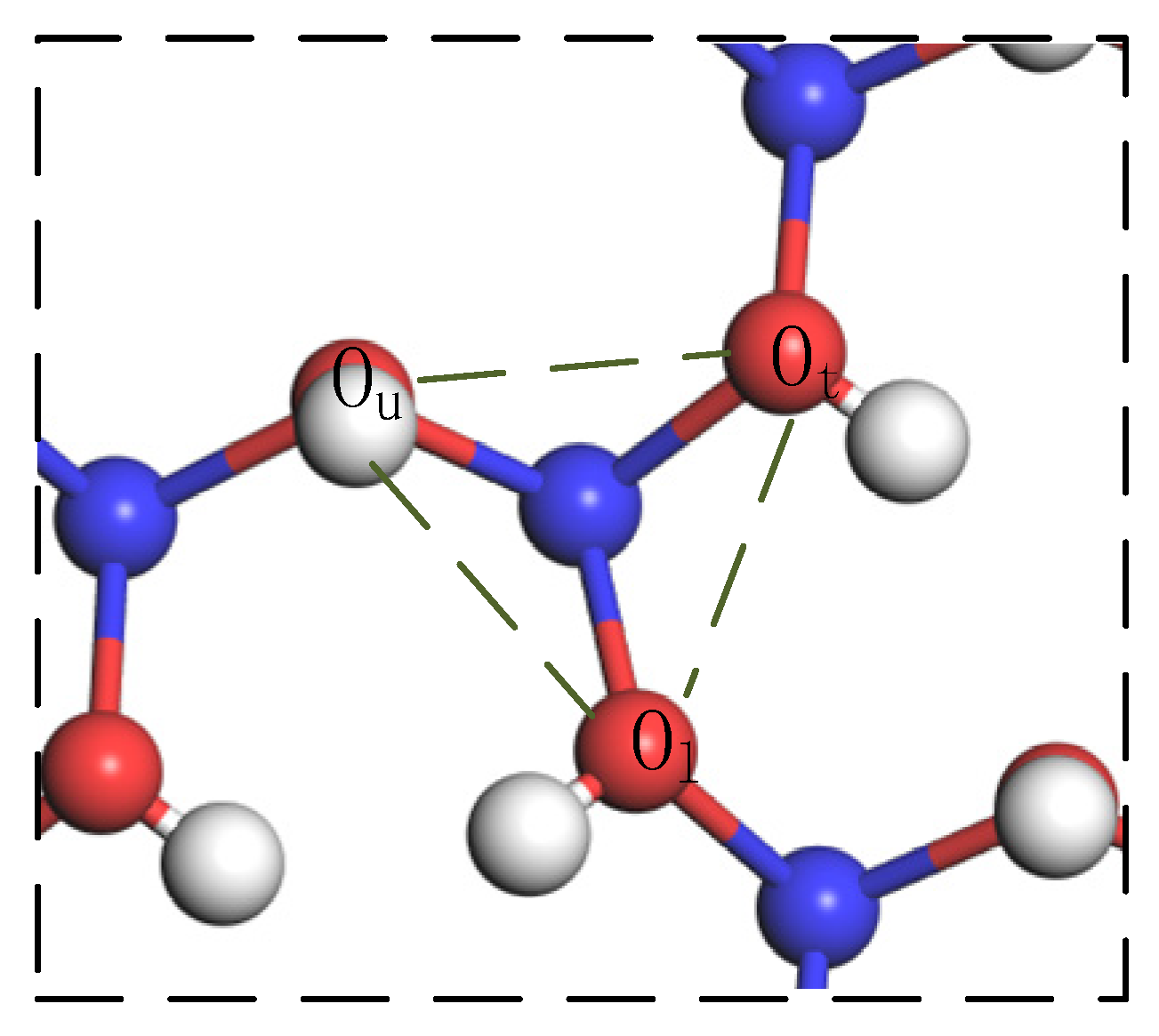
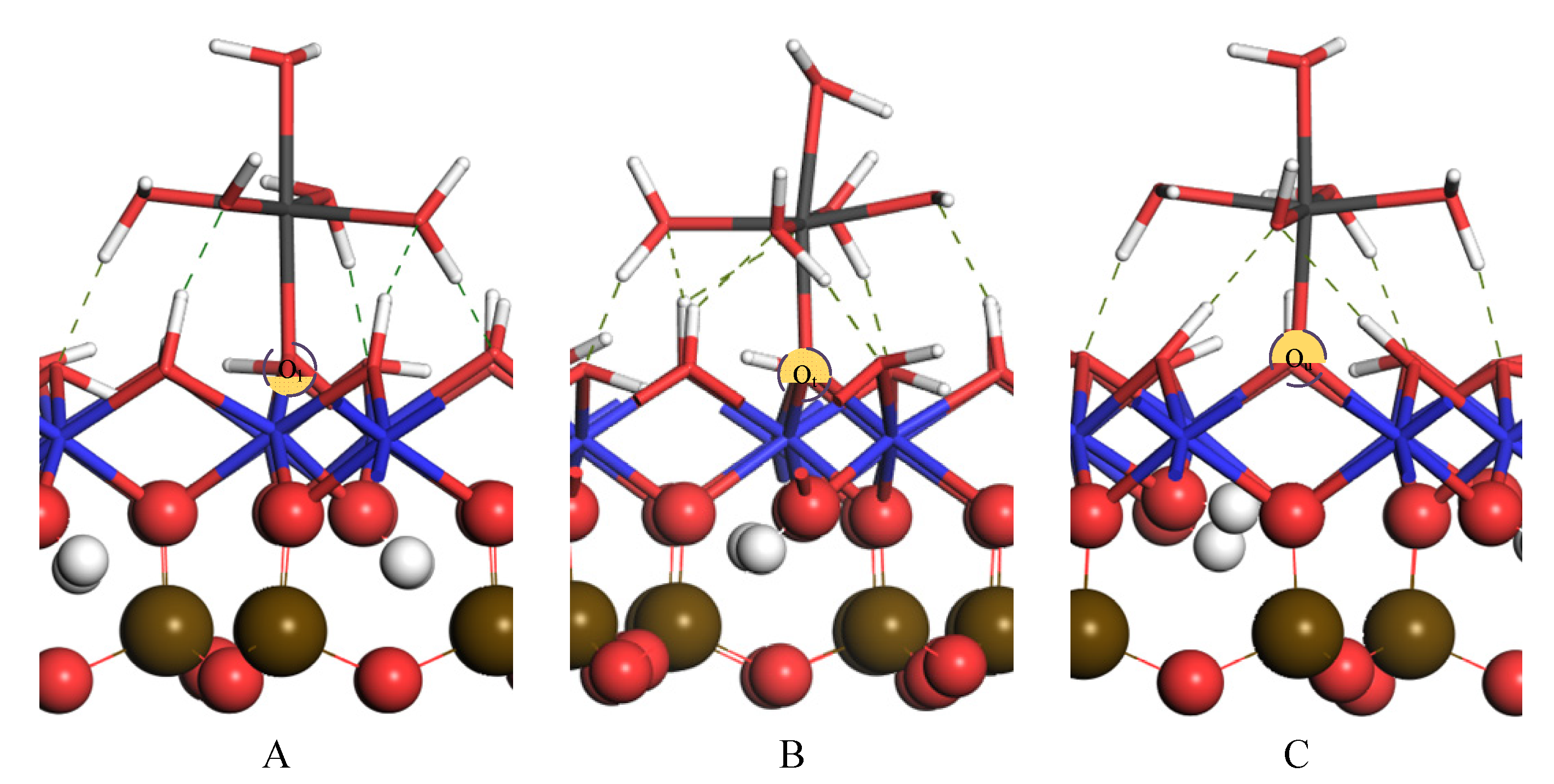
3.3. Inner Layer Adsorption Structure and Analysis
3.3.1. Monodentate Adsorption Structure
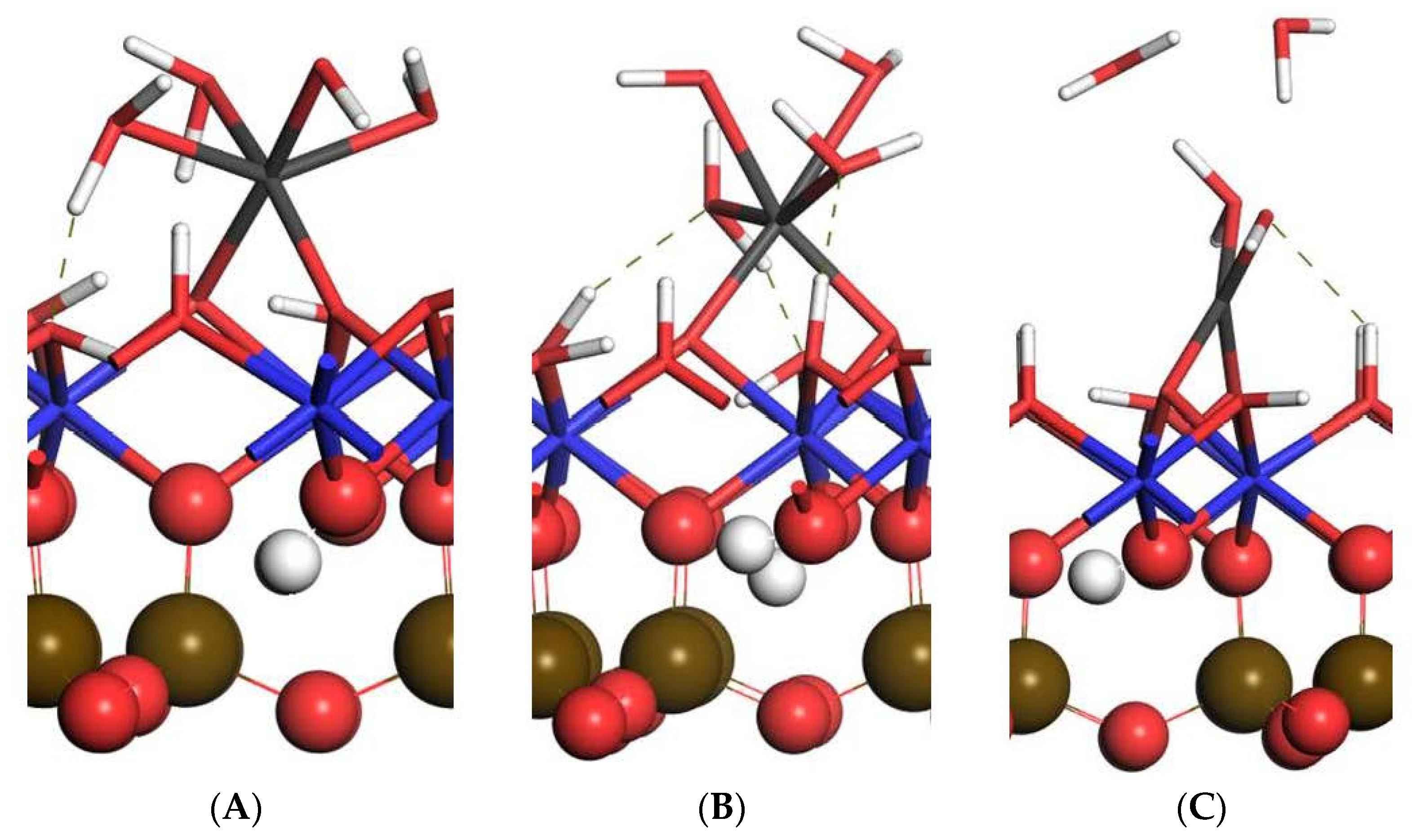
3.3.2. Bidentate Adsorption Structure
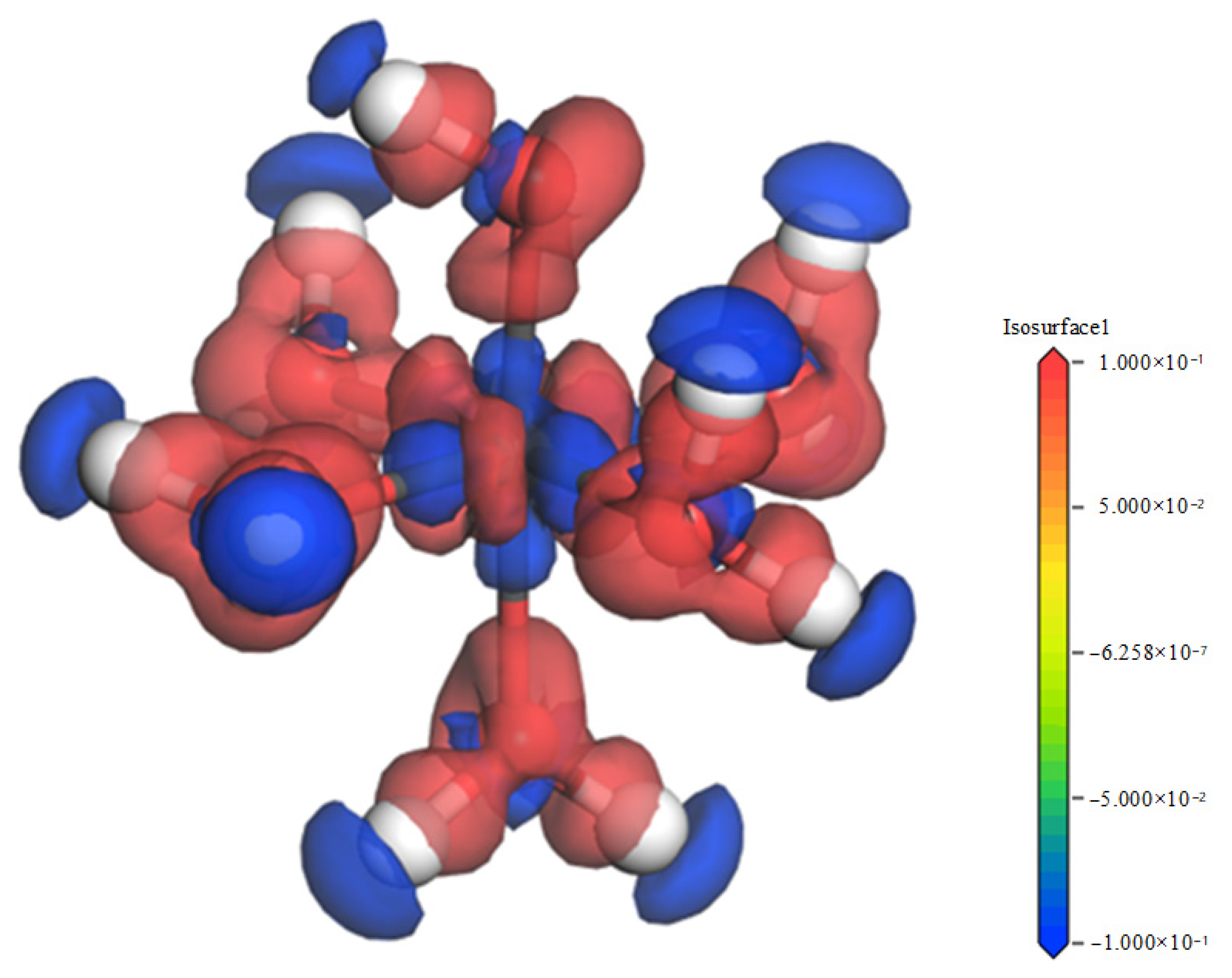
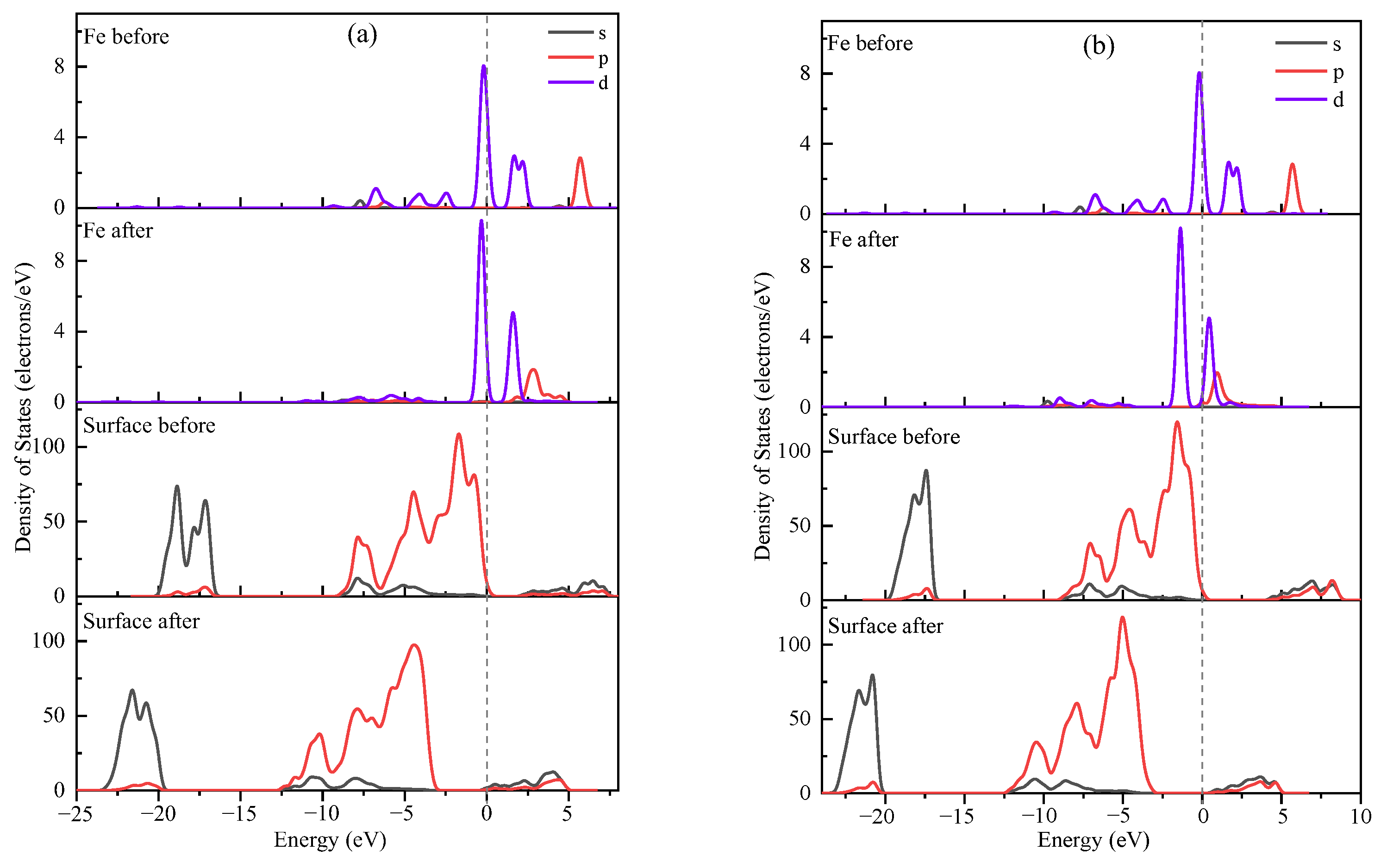
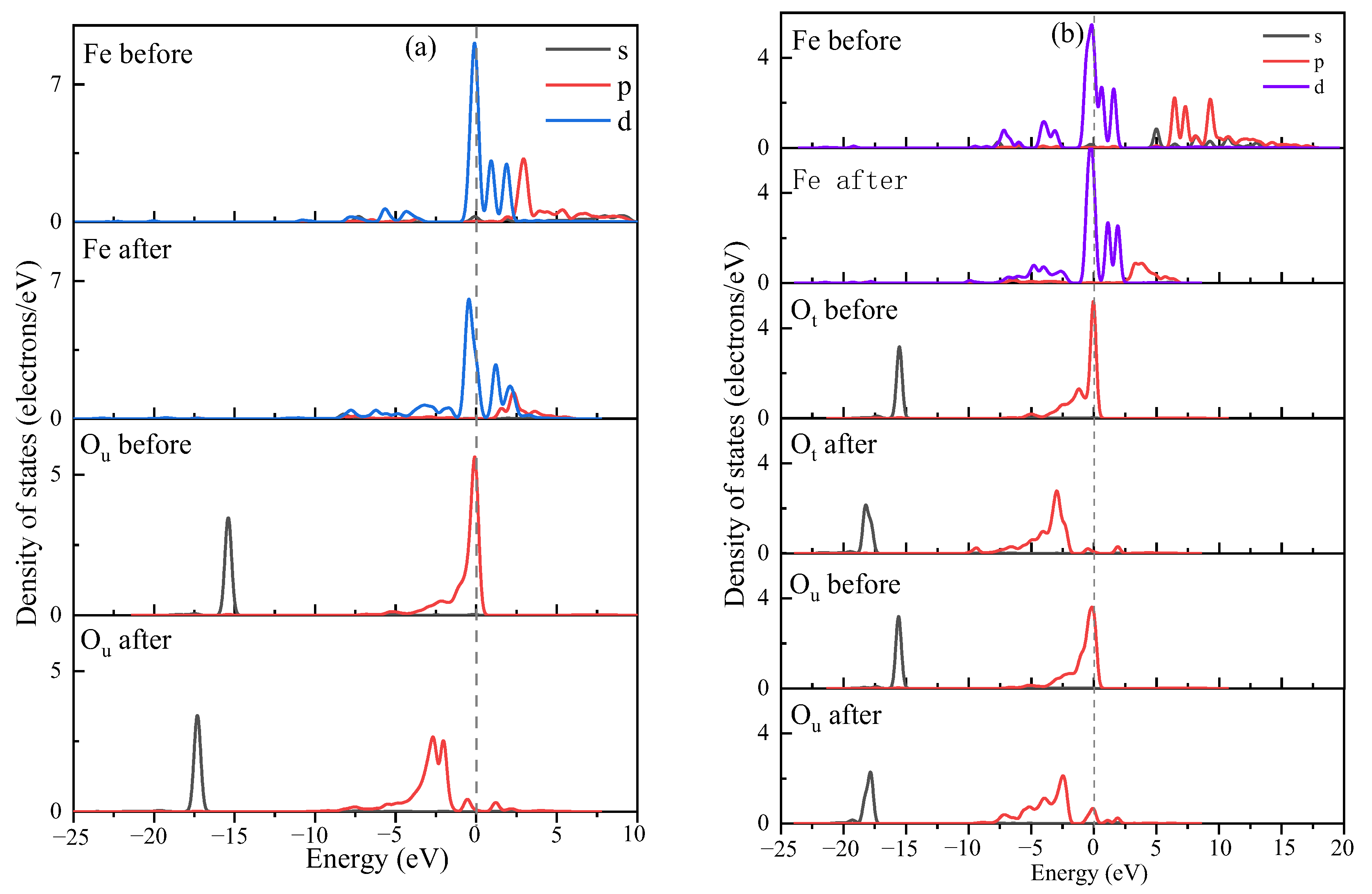
3.4. Explanation of Bonding Mechanisms
4. Conclusions
- (1)
- The stable structure of hydrated Fe(OH)2+ is [Fe(OH)(H2O)5]2+. The adsorbent has a tendency to outer layer adsorb on the kaolinite Si-O surface. If inner layer adsorption occurs on the surface, monodentate adsorption is performed at the upright hydroxyl O and bidentate adsorption is performed between the upright and lying hydroxyl O. Bidentate adsorption has a lower adsorption energy than monodentate adsorption, indicating that adsorbent is more likely to adsorb on the Al-OH surface in the bidentate adsorption manner.
- (2)
- The bonding mechanism and PDOS evaluation confirmed that when outer layer adsorption occurs, the interplay between the adsorbate molecules and the kaolinite molecules was once typically via H-bonding. When outer layer adsorption occurs, the interplay between adsorbate molecules and kaolinite molecules is in the main through vulnerable H-bonds and sturdy chemical bonds, the ionicity of Fe-Os bonds is reduced, and the covalency is enhanced. Compared with monodentate adsorption, bidentate adsorption has lower adsorption energy and greater covalent bonds, so the hydrated Fe(OH)2+ is greater without problems with bidentate adsorbed.
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Huang, C.M.; Wang, C.S. Geochemical features of rare earth elements in process of rock weathering and soil formation. Chin. Rare Earths 2022, 23, 46–49. [Google Scholar]
- Luo, X.P.; Wen, C.J.; Xu, J.; Ma, P.L.; Tang, X.K.; Chi, R.A. Research prowess on and development trend of exploitation technique of ion-absorbed type rare earth ore. Met. Mine 2014, 6, 83–90. [Google Scholar]
- Zhang, H.L.; Xu, Z.J.; Sun, W.; Chen, D.X.; Li, S.; Han, M.J.; Yu, H.; Zhang, C.Y. Selective adsorption mechanism of dodecylamine on the hydrated surface of hematite and quartz. Sep. Purif. Technol. 2021, 275, 119137. [Google Scholar] [CrossRef]
- Wang, X.T.; Chen, Y.R.; Mi, J.Q.; Jiang, J.W.; Jiang, J. Research status of leaching technology and leaching agent of ion-adsorption type rare earth ore. World Nonferrous Met. 2021, 22, 136–139. [Google Scholar]
- Luo, X.P.; Qiu, T.S.; Yan, Q.; Fang, X.H. Research progress and developing orientation of chemical extraction technology of weathing crust strain amass-type rare earth ore. J. Jiangxi Univ. Sci. Technol. 2002, 23, 1–6. [Google Scholar]
- Ouyang, K.S.; Rao, G.H.; Yao, H.Q.; Mao, Y.H. Study of southern RE ore leaching by aluminum inhibition. Rare Met. Cem. Carbides 2003, 31, 1–3. [Google Scholar]
- Qiu, T.S.; Zhu, D.M.; Fang, X.H.; Zeng, Q.H.; Gao, G.K.; Zhu, H.L. Leaching kinetics of ionic rare-earth in ammonia-nitrogen wastewater system added with impurity inhibitors. J. Rare Earths 2014, 32, 1175–1183. [Google Scholar] [CrossRef]
- Qiu, T.S.; Fang, X.H.; Wu, H.Q.; Zeng, Q.H.; Zhu, D.M. Leaching behaviors of iron and aluminum elements of ion-absorbed-rare-earth ore with a new impurity depressant. Trans. Nonferrous Met. Soc. China 2014, 24, 2986–2990. [Google Scholar] [CrossRef]
- Fang, X.H.; Xia, Y.Y.; Qiu, T.S.; Zhu, D.M. Influence of tartaric acid on impurity leaching behavior of ionic rare earth ores. Met. Mine 2018, 6, 94–98. [Google Scholar]
- Fang, X.H.; Zhu, D.M.; Qiu, T.S.; Wu, H.Q. Impurities inhibited leacning of the leach liquor of the weathered crust elution-deposited rare earth ore by adding aluminum inhibitor. Nonferrous Met. Sci. Eng. 2012, 3, 51–55. [Google Scholar] [CrossRef]
- Wu, X.Y.; Zhou, F.; Xu, Y.L.; Feng, J.; Chi, R.A. Research progress on the rare earth leaching agents of weathered crust elution-deposited rare earth ore. Chin. Rare Earths 2021, 42, 109–118. [Google Scholar]
- Zhao, Z.H.; Sang, X.Y.; Zhang, W.B.; Hao, G.H.; Duan, C.K.; Li, D. Application of centrifuging sedimentation on removing aluminum and iron from rare earth solution. Chin. Rare Earths 2007, 28, 95–97. [Google Scholar]
- Xu, Y.H. Removing of aluminum from praseodymium-neodymium carbonate. Hydrometall. China 2005, 24, 92–94. [Google Scholar]
- Yin, J.Q.; Fu, G.M.; Wan, Y.; Tian, J. Development progress of extraction rare earths from the leach liquid of the weathered crust elution-deposited rare earth ore. Jiangxi Sci. 2012, 30, 574–578. [Google Scholar]
- Niyaz, M.M.; Mokhtar, A. Modeling and sensitivity analysis of dyes adsorption onto natural adsorbent from colored textile wastewater. J. Appl. Polym. Sci. 2008, 109, 4043–4048. [Google Scholar]
- Niyaz, M.M.; Mokhtar, A. Numerical finite volume modeling of dye decolorization using immobilized titania nanophotocatalysis. Chem. Eng. J. 2009, 146, 189–193. [Google Scholar]
- Fang, F.; Min, F.; Liu, L.; Chen, J.; Ren, B.; Liu, C. Adsorption of Al(OH)n(3-n)+ (n =2–4) on kaolinite (001) surfaces: A DFT study. Appl. Clay Sci. 2020, 187, 105455. [Google Scholar] [CrossRef]
- Wang, J.; Xia, S.W.; Yu, L.M. Adsorption mechanism of hydrated Pb(OH)+ on the kaolinite (001) surface. Acta Phys. Chim. Sin. 2014, 5, 829–835. [Google Scholar]
- Peng, C.; Min, F.; Liu, L.; Chen, J. The adsorption of CaOH+ on (001) basal and (010) edge surface of Na-montmorillonite: A DFT study: DFT study of adsorption of CaOH+ on (001) Na-montmorillonite surface. Surf. Interface Anal. 2017, 49, 267–277. [Google Scholar] [CrossRef]
- Zhang, Z.J.; Zhou, Q.; Yuan, Z.T.; Zhao, L.; Dong, J.D. Adsorption of Mg2+ and K+ on the kaolinite (0 0 1) surface in aqueous system: A combined DFT and AIMD study with an experimental verification. Appl. Surf. Sci. 2021, 538, 148158. [Google Scholar] [CrossRef]
- Qiu, T.S.; Qiu, S.; Wu, H.; Yan, H.S.; Li, X.B.; Zhou, X.W. Adsorption of hydrated [Y(OH)2]+ on kaolinite (001) surface: Insight from DFT simulation. Powder Technol. 2021, 387, 80–87. [Google Scholar] [CrossRef]
- Qiu, S.; Wu, H.; Yan, H.S.; Li, X.B.; Zhou, X.W.; Qiu, T.S. Theoretical investigation of hydrated [Lu(OH)2]+ adsorption on kaolinite (001) surface with DFT calculations. Appl. Surf. Sci. 2021, 565, 150473. [Google Scholar] [CrossRef]
- Miao, Y.Q.; Yan, H.S.; Qiu, X.H.; Zhou, X.W.; Zhu, D.M.; Li, X.B.; Qiu, T.S. Adsorption of hydrated Al3+ on the kaolinite (001) surface: A density functional theory study. Appl. Clay Sci. 2022, 225, 106498. [Google Scholar] [CrossRef]
- Yan, H.S. First-Principles Study on the Adsorption of Hydrated Rare Earth Ions on the Surface of Kaolinite; Jiangxi University of Science and Technology: Ganzhou, China, 2019. [Google Scholar]
- Liu, X.D.; Jan Meijer, E.; Lu, X.C.; Wang, R.C. First-principles molecular dynamics insight into Fe2+ compounds adsorbed on edge surface of clay minerals. Clays Clay Miner. 2012, 60, 341–347. [Google Scholar] [CrossRef]
- Feng, X.; Onel, O.; Council-Troche, M.; MacCormac, B.L.; Noble, A.; Yoon, R.H.; Morris, J.R. Rare earth ion-adsorption clays in the presence of iron at basic pH: Adsorption mechanism and extraction method. Appl. Clay Sci. 2023, 231, 106744. [Google Scholar] [CrossRef]
- Hohenberg, P.; Kohn, W. Inhomogeneous electron gas. Phys. Rev. J. 1964, 136, B864–B871. [Google Scholar] [CrossRef] [Green Version]
- Kohn, W.; Sham, L.J. Self-consistent equations including exchange and correlation effects. Phys. Rev. J. 1965, 140, A1133–A1138. [Google Scholar] [CrossRef] [Green Version]
- Clark, S.J.; Segall, M.D.; Pickard, C.J.; Hasnip, P.J.; Probert, M.J.; Refson, K.; Payne, M.C. First principles methods using CASTEP. Z. Fur Krist. Cryst. Mater. 2005, 220, 567–570. [Google Scholar] [CrossRef] [Green Version]
- Wu, Z.; Cohen, R.E. More accurate generalized gradient approximation for solids. Phys. Rev. B 2006, 73, e235116. [Google Scholar] [CrossRef] [Green Version]
- Ireta, J.; Neugebauer, J.; Scheffler, M. On the accuracy of DFT for describing H-bonds: Dependence on the bond directionality. J. Phys. Chem. A 2004, 108, 5692–5698. [Google Scholar] [CrossRef] [Green Version]
- Vanderbilt, D. Soft self-consistent pseudopotentials in a generalized eigenvalue formalism. Phys. Rev. B 1990, 41, 7892–7895. [Google Scholar] [CrossRef] [PubMed]
- Monkhorst, H.J.; Pack, J.D. Special points for brillouin-zone integrations. Phys. Rev. B 1976, 13, 5188–5192. [Google Scholar] [CrossRef]
- Pack, J.D.; Monkkorst, H.J. Special points for brilliouin-zone zategrations—A reply. Phys. Rev. B 1976, 16, 1748–1749. [Google Scholar] [CrossRef]
- Yin, Z.G.; Hu, Y.H.; Sun, W.; Zhang, C.Y.; He, J.Y.; Xu, Z.J.; Zou, J.X.; Guan, C.P.; Zhang, C.H.; Guan, Q.J.; et al. Adsorption mechanism of 4-amino-5-mercapto-1,2,4-triazole as flotation reagent on chalcopyrite. Langmuir 2018, 34, 4071–4083. [Google Scholar] [CrossRef]
- He, J.Y.; Zhang, H.L.; Yue, T.; Sun, W.; Hu, Y.H.; Zhang, C.Y. Effects of hydration on the adsorption of benzohydroxamic acid on the lead-ion-activated cassiterite surface: A DFT study. Langmuir 2021, 37, 2205–2212. [Google Scholar] [CrossRef]
- Zhang, C.; Zhang, Q.J.; Huang, B. Contamination prevention control for ion-adsorbed rare earth deposit mining. Energy Sav. Non Ferr. Metall. 2021, 37, 46–49. [Google Scholar]
- Zhang, H.L.; Sun, W.; Zhu, Y.G.; He, J.Y.; Chen, D.X.; Zhang, C.Y. Effects of the goethite surface hydration microstructure on the adsorption of the collectors dodecylamine and sodium oleate. Langmuir 2021, 37, 10052–10060. [Google Scholar] [CrossRef]
- He, J.Y.; Wang, L.; Zhang, C.Y.; Sun, W.; Yin, Z.G.; Zhang, H.L.; Chen, D.X.; Pei, Y. A high throughput screening model of solidophilic flotation reagents for chalcopyrite based on quantum chemistry calculations and machine learning. Miner. Eng. 2022, 177, 107375. [Google Scholar] [CrossRef]
- Chen, J.H. The interaction of flotation reagents with metal ions in mineral surfaces: A perspective from coordination chemistry. Miner. Eng. 2021, 171, 107067. [Google Scholar] [CrossRef]
- Luo, A.R.; Chen, J.H. Effect of hydration and hydroxylation on the adsorption of metal ions on quartz surfaces: DFT study. Appl. Surf. Sci. 2022, 595, 153553. [Google Scholar] [CrossRef]
- Wang, X.Y.; Liu, W.G.; Duan, H.; Wang, B.Y.; Han, C.; Wei, D.Z. The adsorption mechanism of calcium ion on quartz (101) surface: A DFT study. Power Technol. 2018, 329, 158–166. [Google Scholar] [CrossRef]
- Mulliken, R.S. Electronic population analysis on LCAO–MO molecular wave functions. J. Chem. Phys. 1955, 23, 1833–1840. [Google Scholar] [CrossRef] [Green Version]
- Miao, Y.Q.; Yan, H.S.; Hong, B.G.; Zhou, X.W.; Tong, L.C.; Xiao, Y.F.; Qiu, T.S. DFT study of the effect of impurity defects on the inner-layer adsorption of hydrated Al(OH)2+ on the kaolinite (0 0 1) surface. J. Mol. Liq. 2022, 368, 120819. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| n | R (Fe-Ooh)/Å a | R (Fe-Ow)min/Å | R (Fe-Ow)max b/Å | R (Fe-Ow)mean c/Å | QFe d | Ebind |
|---|---|---|---|---|---|---|
| 1 | 1.693 | 1.847 | 1.847 | 1.847 | 1.62 | −338.79 |
| 2 | 1.720 | 1.878 | 1.881 | 1.880 | 1.52 | −924.35 |
| 3 | 1.725 | 1.863 | 1.896 | 1.884 | 1.39 | −3043.14 |
| 4 | 1.761 | 1.901 | 1.969 | 1.929 | 1.40 | −2996.45 |
| 5 | 1.772 | 1.940 | 1.972 | 1.957 | 1.40 | −3215.26 |
| 6 | 1.777 | 1.940 | 3.567 | 2.223 | 1.39 | −3321.96 |
| Atom | s | p | d | Total | Charge/e |
|---|---|---|---|---|---|
| O | 1.90 | 4.86 | 0 | 6.76 | −0.76 |
| O | 1.86 | 5.03 | 0 | 6.89 | −0.89 |
| O | 1.85 | 5.06 | 0 | 6.91 | −0.91 |
| O | 1.85 | 5.09 | 0 | 6.94 | −0.94 |
| O | 1.86 | 5.04 | 0 | 6.90 | −0.90 |
| O | 1.85 | 5.06 | 0 | 6.91 | −0.91 |
| Fe | 0.22 | −0.07 | 6.45 | 6.60 | 1.40 |
| Form | M a | (Fe-Ooh) b | (Fe-Ow)mean c | Os-Hw | Ow-Hs | Eads |
|---|---|---|---|---|---|---|
| P | 5 | 1.943 | 1.989 | 1.750, 1.780, 1.802 2.150, 2.438, 2.541 2.551 | / | −1163.86 |
| Q | 5 | 1.946 | 1.993 | 1.545, 2.202 | 1.704, 2.363 | −993.82 |
| Surface | State | s | p | d | Total | Charge/e |
|---|---|---|---|---|---|---|
| Si-O | Fe before | 0.22 | −0.07 | 6.45 | 6.60 | 1.40 |
| Fe after | 0.22 | −0.12 | 6.73 | 6.87 | 1.13 | |
| Al-OH | Fe before | 0.22 | −0.07 | 6.45 | 6.60 | 1.40 |
| Fe after | 0.21 | 0.01 | 6.74 | 6.97 | 1.03 |
| Form | M a | Fe-Os b | Fe-Ooh c | Fe-Ow d | Os-Hw | Ow-Hs | Eads |
|---|---|---|---|---|---|---|---|
| P | 5 | \ | 1.943 | 1.960, 1.965, 2.002 2.006, 2.013 | 1.750, 1.780, 1.802 2.150, 2.438, 2.541 2.551 | / | −1163.86 |
| Q | 5 | \ | 1.946 | 1.973, 1.980, 1.994 2.008, 2.008 | 1.545, 2.202 | 1.704, 2.363 | −993.82 |
| A | 4 | 2.021 | 1.952 | 2.005, 2.008 2.024, 2.044 | 1.519, 1.531, 1.705 | 1.473, 1.862 | −1339.67 |
| B | 4 | 2.011 | 1.984 | 1.968, 1.979 2.017, 2.030 | 1.495, 1.502, 1.504 | 1.698, 1.803 1.812, 2.448 | −1384.67 |
| C | 4 | 2.022 | 1.966 | 1.997, 2.025 2.027, 2.030 | 1.467, 1.550, 1.618 | 1.648, 1.863 | −1423.83 |
| D | 3 | 1.851 2.032 | 1.945 | 1.949, 1.994, 2.012 | 1.400 | \ | −1713.89 |
| E | 3 | 1.859 1.919 | 2.058 | 1.922, 2.046, 2.233 | 1.289 | 1.618, 1.951 | −1598.90 |
| F | 1 | 1.753 1.837 | 1.804 | 1.936 | \ | 2.074 | −1664.17 |
| States | Fe | Os | Fe-Os | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| s | p | d | Total | Charge/e | s | p | Total | Charge/e | ||
| Before | 0.20 | −0.04 | 6.44 | 6.60 | 1.40 | 1.91 | 4.90 | 6.81 | −0.81 | / |
| After | 0.24 | −0.00 | 6.75 | 6.99 | 1.01 | 1.85 | 5.19 | 7.04 | −1.04 | 0.32 |
| Charge | 0.04 | 0.04 | 0.31 | 0.39 | −0.39 | −0.06 | 0.29 | 0.23 | −0.23 | / |
| Form | Qo | QH | Qo′ | QH′ | Qo a | ΔQH |
|---|---|---|---|---|---|---|
| C | −4.36 | 4.95 | −4.46 | 3.83 | −0.10 | −1.12 |
| D | −3.34 | 3.95 | −3.48 | 2.93 | −0.14 | −1.02 |
| States | Fe | Ol | Ou | Fe-Os | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| s | p | d | Total | Charge/e | s | p | Total | Charge/e | s | p | Total | Charge/e | ||
| Before | 0.18 | −0.01 | 6.44 | 6.61 | 1.39 | 1.91 | 4.95 | 6.86 | −0.86 | 1.90 | 4.98 | 6.88 | −0.88 | 0.24 0.36 |
| After | 0.23 | −0.04 | 6.51 | 6.70 | 1.30 | 1.87 | 5.13 | 7.00 | −1.00 | 1.86 | 5.07 | 6.93 | −0.93 | |
| Charge | 0.05 | −0.03 | 0.07 | 0.09 | −0.09 | −0.04 | 0.18 | 0.14 | −0.14 | −0.04 | 0.09 | 0.05 | −0.05 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, H.; Miao, Y.; Li, Y.; Yan, H.; Tan, J.; Qiu, S.; Wu, H.; Qiu, T. Density Functional Theory Study on the Adsorption of Fe(OH)2+ on Kaolinite Surface in Water Environment. Processes 2023, 11, 38. https://doi.org/10.3390/pr11010038
Wu H, Miao Y, Li Y, Yan H, Tan J, Qiu S, Wu H, Qiu T. Density Functional Theory Study on the Adsorption of Fe(OH)2+ on Kaolinite Surface in Water Environment. Processes. 2023; 11(1):38. https://doi.org/10.3390/pr11010038
Chicago/Turabian StyleWu, Hongqiang, Yuqi Miao, Yong Li, Huashan Yan, Jinbiao Tan, Sen Qiu, Hao Wu, and Tingsheng Qiu. 2023. "Density Functional Theory Study on the Adsorption of Fe(OH)2+ on Kaolinite Surface in Water Environment" Processes 11, no. 1: 38. https://doi.org/10.3390/pr11010038