
Machine Learning Prediction of Critical Temperature of Organic Refrigerants by Molecular Topology

Abstract
:1. Introduction
2. Methods
2.1. ML Algorithms
2.1.1. Support Vector Regression
2.1.2. Decision Tree
2.1.3. Random Forest
2.1.4. Multilayer Perceptron
2.2. Datasets
2.3. Feature Extraction and Data Preprocessing
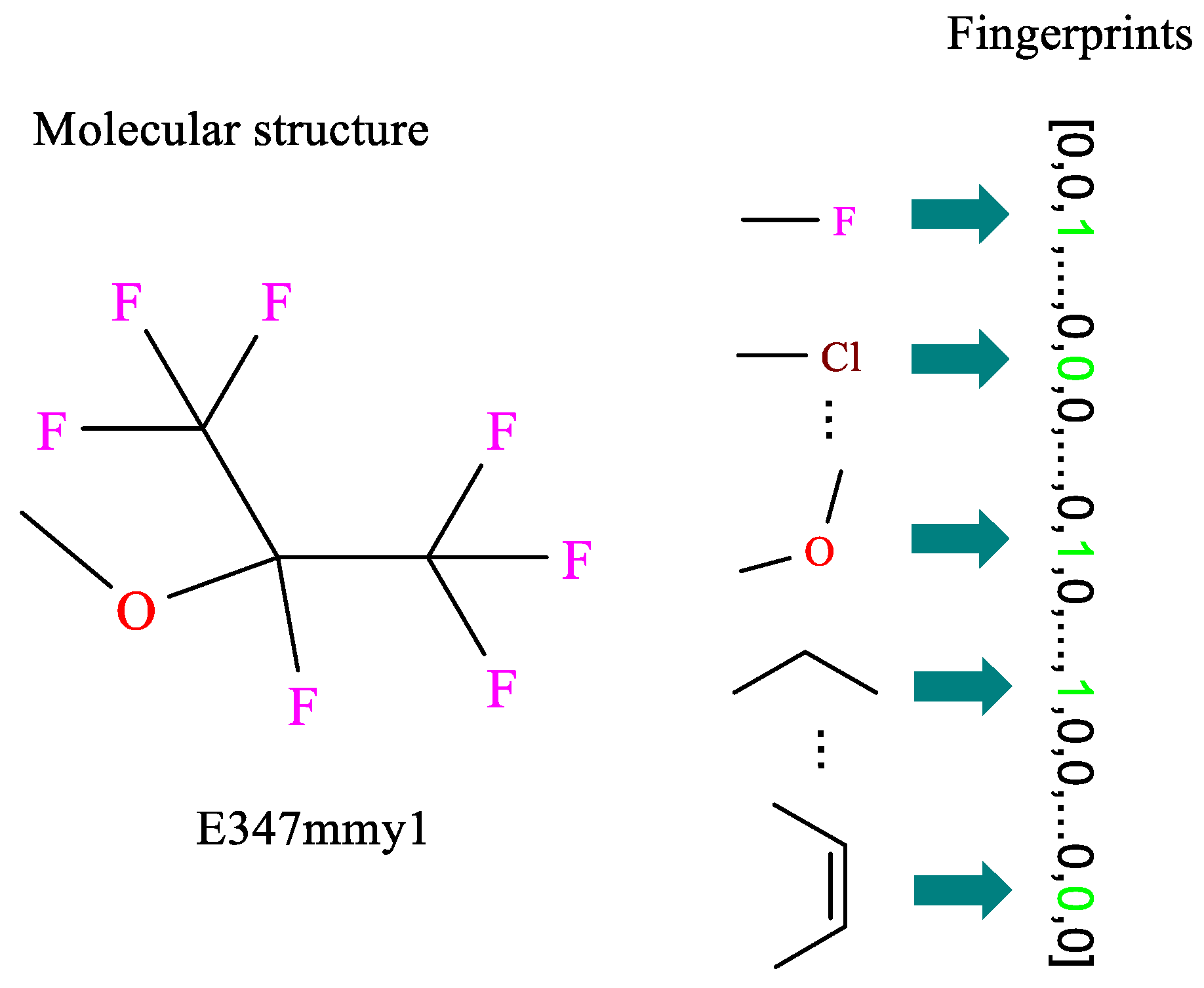
2.3.1. Molecular Fingerprints
2.3.2. Topological Index
2.4. Model Validation
3. Results and Discussion
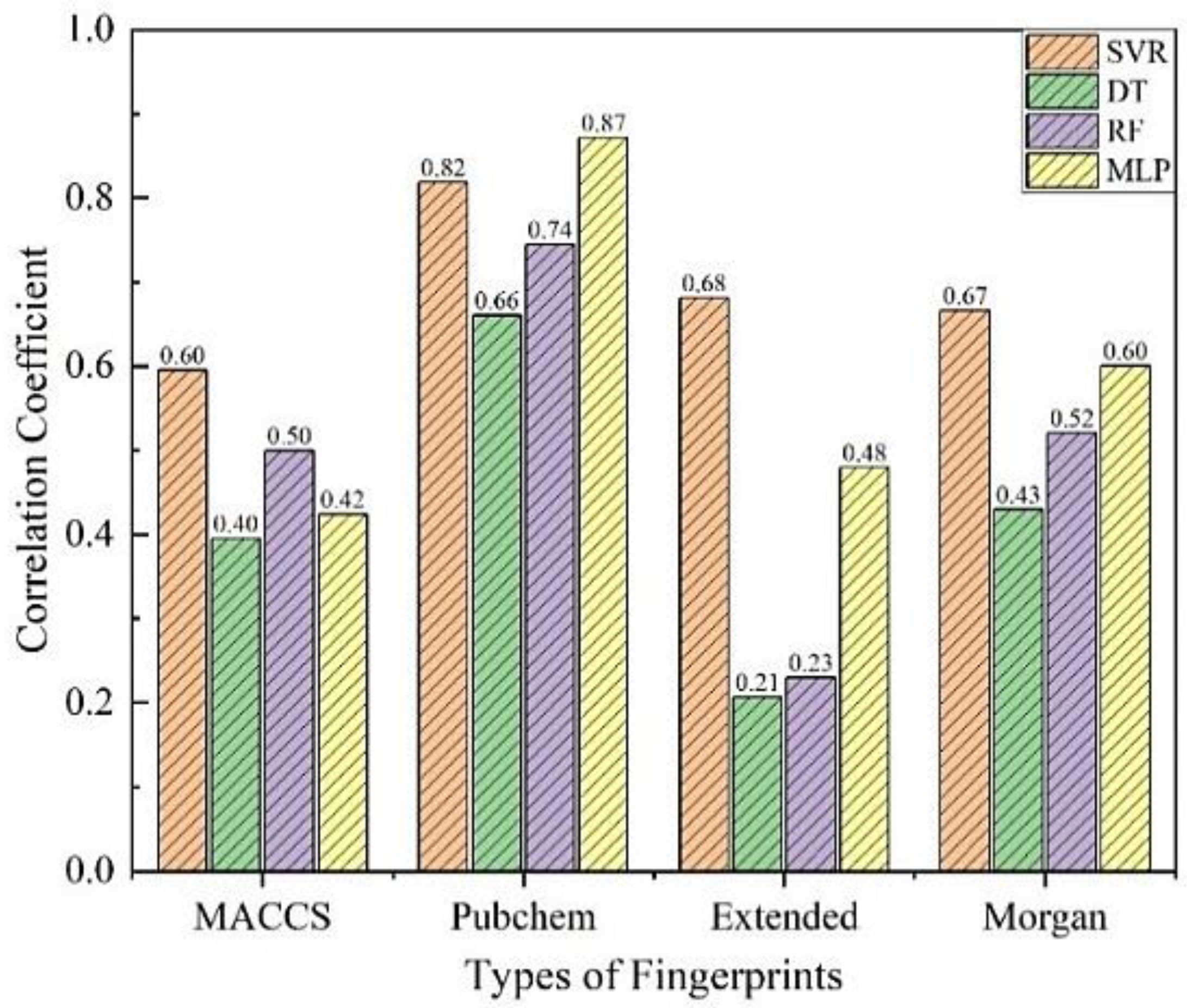
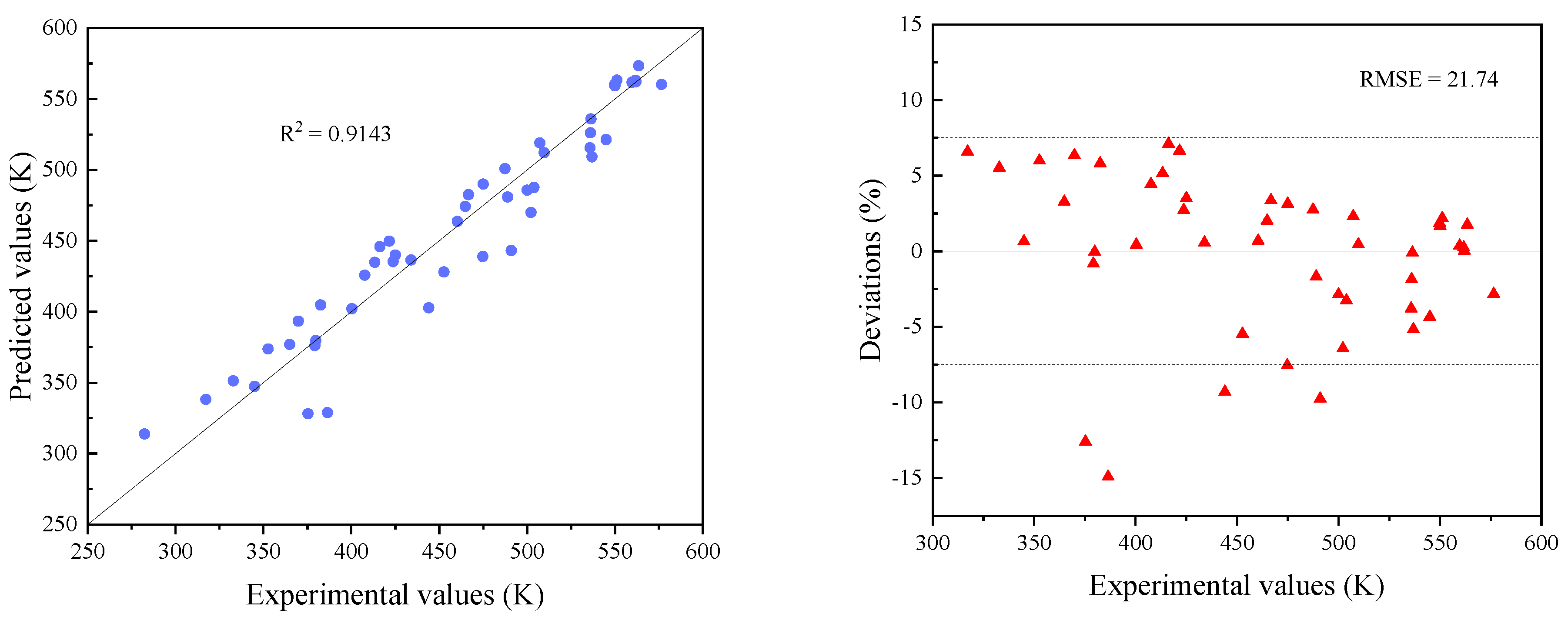
3.1. Preliminary Screening of Models
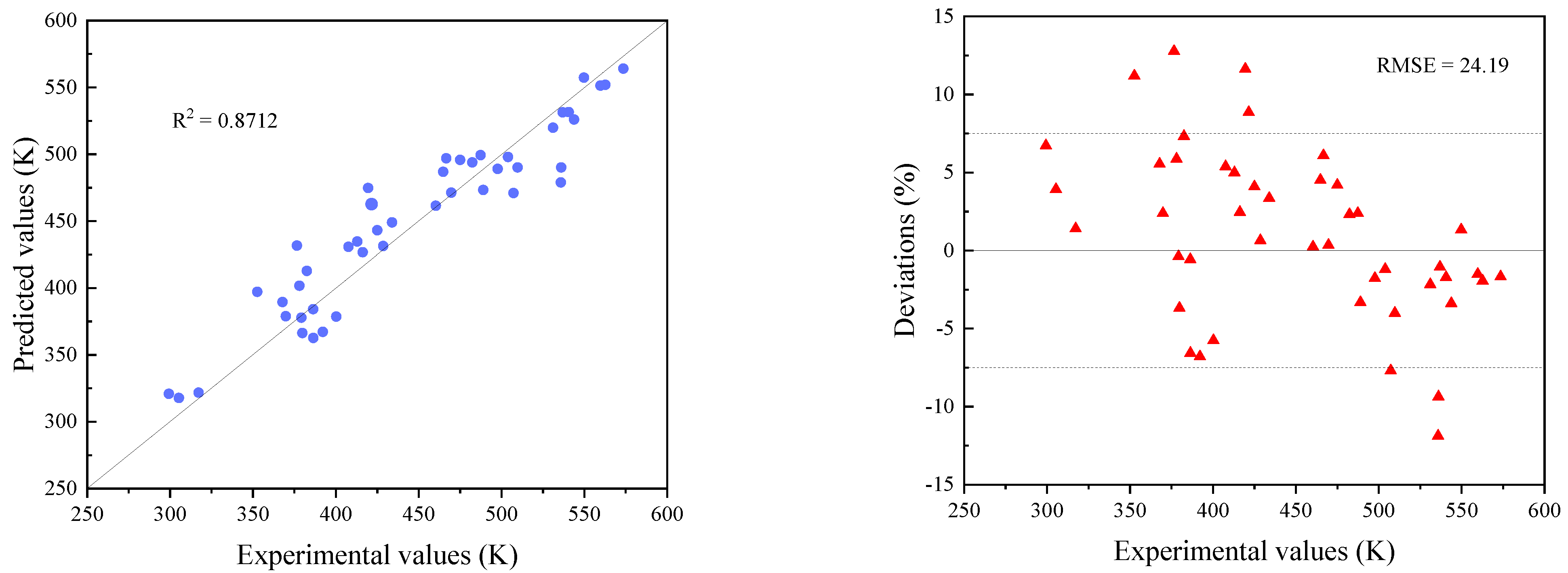
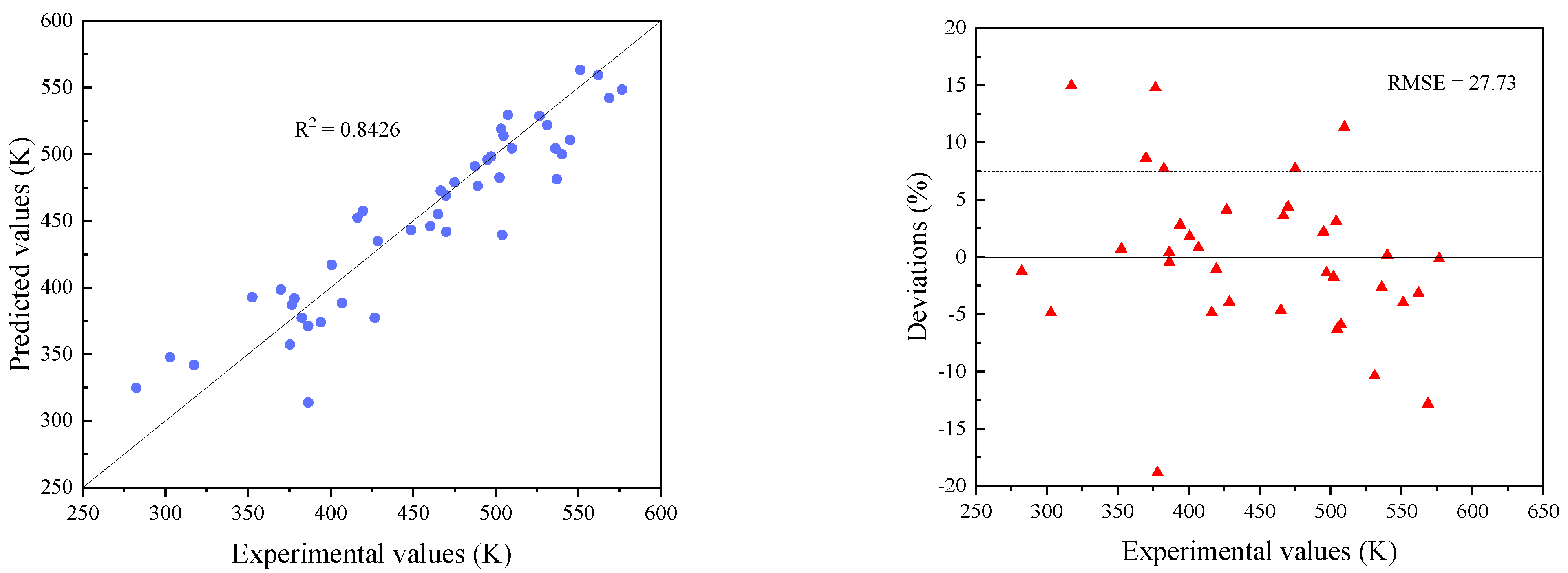
3.2. Modification of Models
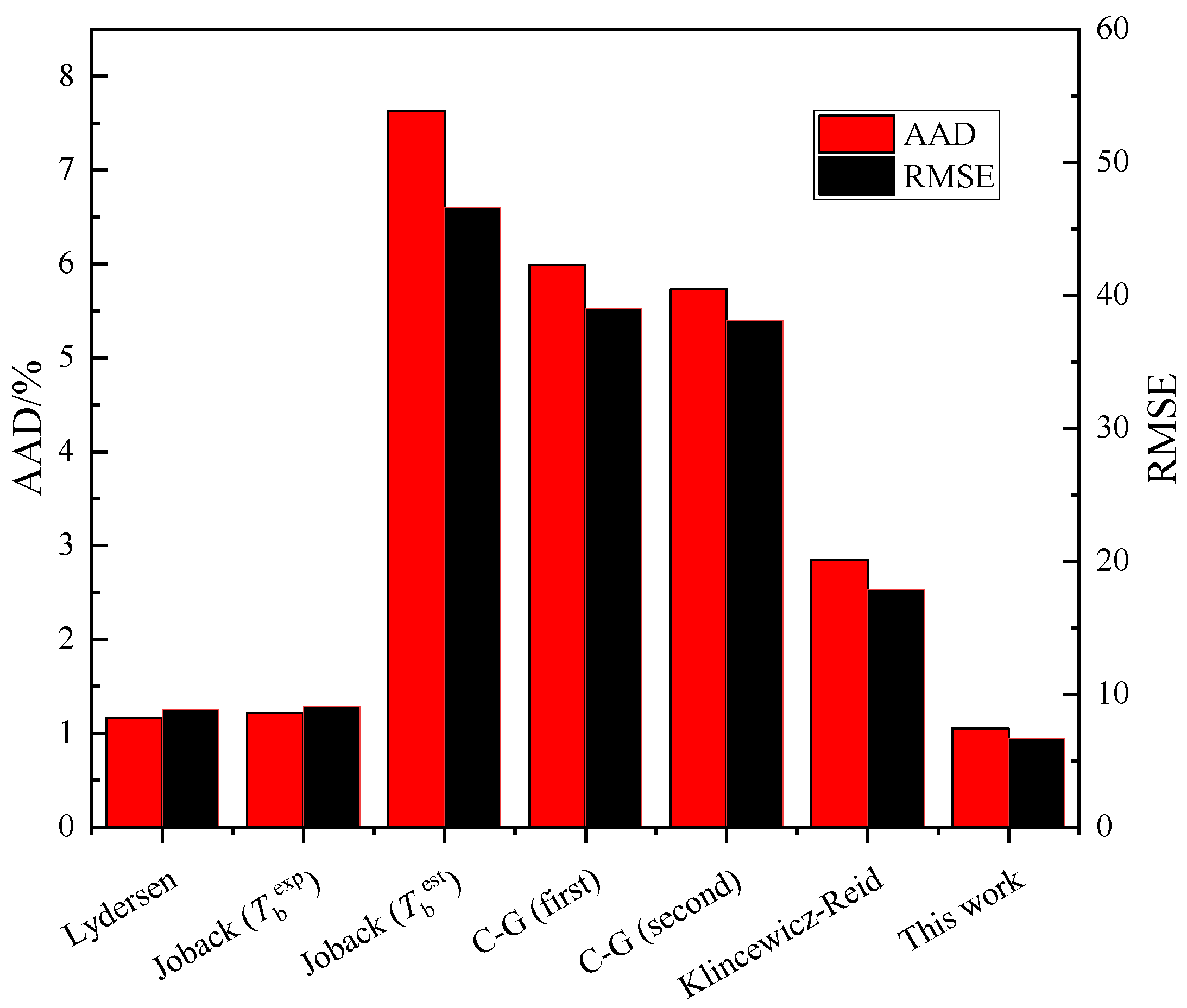
3.3. Comparisons with Existing Methods
3.4. Distinction of Isomers
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Nomenclature
| AAD | absolute average deviation |
| DT | decision tree |
| GCM | group contribution method |
| GWP | global warming potential |
| ML | machine learning |
| MLP | multilayer perceptron |
| MF | molecular fingerprint |
| ODP | ozone depletion potential |
| ORC | organic Rankine cycle |
| QSPR | quantitative structure property relationship |
| R2 | coefficient of determination |
| RMSE | root mean square error |
| RF | random forest |
| SMILES | simplified molecular input line entry specification |
| SVR | support vector regression |
| TI | topological index |
References
- Wang, X.; Li, X.; Li, Q.; Liu, L.; Liu, C. Performance of a solar thermal power plant with direct air-cooled supercritical carbon dioxide Brayton cycle under off-design conditions. Appl. Energy 2020, 261, 114359. [Google Scholar] [CrossRef]
- Wang, S.; Liu, C.; Zhang, C.; Xu, X.; Li, Q. Thermo-economic evaluations of dual pressure organic Rankine cycle (DPORC) driven by geothermal heat source. J. Renew. Sustain. Energy 2018, 10, 063901. [Google Scholar] [CrossRef]
- Chen, X.; Liu, C.; Li, Q.; Wang, X.; Wang, S. Dynamic behavior of supercritical organic Rankine cycle using zeotropic mixture working fluids. Energy 2020, 191, 116576. [Google Scholar] [CrossRef]
- Wang, S.; Liu, C.; Li, Q.; Liu, L.; Zhang, C. Selection principle of working fluid for organic Rankine cycle based on environmental benefits and economic performance. Appl. Therm. Eng. 2020, 178, 115598. [Google Scholar] [CrossRef]
- Mahmoudi, A.; Fazli, M.; Morad, M.R. A recent review of waste heat recovery by Organic Rankine Cycle. Appl. Therm. Eng. 2018, 143, 660–675. [Google Scholar] [CrossRef]
- Samudra, A.; Sahinidis, N.V. Design of Secondary Refrigerants: A Combined Optimization-Enumeration Approach. In Design for Energy and the Environment; El Halwagi, M.M., Linninger, A.A., Eds.; CRC Press: Boca Raton, FL, USA, 2010; pp. 879–886. [Google Scholar]
- Liu, B.T.; Chien, K.H.; Wang, C.C. Effect of working fluids on organic Rankine cycle for waste heat recovery. Energy 2004, 29, 1207–1217. [Google Scholar] [CrossRef]
- Kay, W.B.; Pak, S.C. Determination of the critical constants of high-boiling hydrocarbons Experiments with gallium as a containing liquid. J. Chem. Thermodyn. 1980, 12, 673–681. [Google Scholar] [CrossRef]
- Kleinrahm, R.; Wagner, W. Measurement and correlation of the equilibrium liquid and vapour densities and the vapour pressure along the coexistence curve of methane. J. Chem. Thermodyn. 1986, 18, 739–760. [Google Scholar] [CrossRef]
- Su, W.; Zhao, L.; Deng, S. Group contribution methods in thermodynamic cycles: Physical properties estimation of pure working fluids. Renew. Sustain. Energy Rev. 2017, 79, 984–1001. [Google Scholar] [CrossRef]
- Reid, R.C.; Sherwood, T.K.; Street, R.E. The properties of gases and liquids. Phys. Today 1959, 18, 739–760. [Google Scholar] [CrossRef]
- Vejahati, F.; Nikoo, M.B.; Mokhatab, S.; Towler, B.F. Simple Correlation Estimates Critical Properties of Alkanes. Pet. Sci. Technol. 2007, 25, 1115–1123. [Google Scholar] [CrossRef]
- Klincewicz, K.M.; Reid, R.C. Estimation of critical properties with group contribution methods. AIChE J. 1984, 30, 137–142. [Google Scholar] [CrossRef]
- Riedel, L. Additives Verfahren zur Abschätzung der kritischen Temperatur aus dem normalen Siedepunkt. Chem. Ing. Tech. 1952, 24, 353–357. [Google Scholar] [CrossRef]
- Lydersen, L.A. Estimation of Critical Properties of Organic Compounds Vol. 2; Engineering Experiment Station Report 3; College of Engineering, University of Wisconsin: Madison, WI, USA, 1955; p. 12S. [Google Scholar]
- Joback, K.G.; Reid, R.C. Estimation of pure-component properties from group-contribution. Chem. Eng. Commun. 1987, 57, 233–243. [Google Scholar] [CrossRef]
- Constantinou, L.; Gani, R. New group contribution method for estimating properties of pure compounds. AIChE J. 1994, 40, 1697–1710. [Google Scholar] [CrossRef]
- Marrero-Morejón, J.; Pardillo-Fontdevila, E. Estimation of pure compound properties using group-interaction contributions. AIChE J. 1999, 45, 615–621. [Google Scholar] [CrossRef]
- Wang, Q.; Ma, P.; Jia, Q.; Xia, S. Position Group Contribution Method for the Prediction of Critical Temperatures of Organic Compounds. J. Chem. Eng. Data 2008, 53, 1103–1109. [Google Scholar] [CrossRef]
- Frenkel, D.; Smit, B. Understanding Molecular Simulation: From Algorithms to Applications, 2nd ed.; Academic Press: New York, NY, USA, 2002. [Google Scholar]
- Gabriele, R. Molecular Simulation Studies on the Vapor–Liquid Phase Equilibria of Binary Mixtures of R-1234yf and R-1234ze(E) with R-32 and CO2. J. Chem. Eng. Data 2013, 58, 1867–1873. [Google Scholar]
- Dong, X.; Gong, M.; Li, X.; Wu, J.; Yang, Z. Molecular modeling and simulation of vapor–liquid equilibrium of the refrigerant R152a and its mixture R152a+R32. Fluid Phase Equilibria 2015, 394, 93–100. [Google Scholar]
- Cai, S.; Li, Q.; Liu, C.; Zhou, Y. Evaporation of R32/R152a mixtures on the Pt surface: A molecular dynamics study. Int. J. Refrig. 2020, 113, 156–163. [Google Scholar] [CrossRef]
- Li, Q.; Xiao, Y.; Shi, X.; Song, S. Rapid Evaporation of Water on Graphene/Graphene-Oxide: A Molecular Dynamics Study. Nanomaterials 2017, 7, 265. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Liu, C.; Li, Q.; Shi, X. Molecular simulation of thermal energy storage of mixed CO2/IRMOF-1 nanoparticle nanofluid. Int. J. Heat Mass Transf. 2018, 125, 1345–1348. [Google Scholar] [CrossRef]
- Huo, E.; Liu, C.; Xu, X.; Li, Q.; Dang, C. The oxidation decom position mechanisms of HFO-1336mzz(Z) as an environmentally friendly refrigerant in O2/H2O environment. Energy 2019, 185, 1154–1162. [Google Scholar] [CrossRef]
- Maleki, A.A.H.; Mahariq, I. Machine learning-based approaches for modeling thermophysical properties of hybrid nanofluids: A comprehensive review. J. Mol. Liq. 2021, 322, 114843. [Google Scholar] [CrossRef]
- Mitchell, J. Machine learning methods in chemoinformatics. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2014, 4, 468–481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Venkatraman, V.; Evjen, S.; Knuutila, H.K.; Fiksdahl, A.; Alsberg, B.K. Predicting ionic liquid melting points using machine learning. J. Mol. Liq. 2018, 264, 318–326. [Google Scholar] [CrossRef]
- Deng, S.; Su, W.; Zhao, L. A neural network for predicting normal boiling point of pure refrigerants using molecular groups and a topological index. Int. J. Refrig. 2016, 63, 63–71. [Google Scholar] [CrossRef]
- Zolfaghari, H.; Yousefi, F. Thermodynamic properties of lubricant/refrigerant mixtures using statistical mechanics and artificial intelligence. Int. J. Refrig. 2017, 80, 130–144. [Google Scholar] [CrossRef]
- Gao, N.A.; Wang, X.B.; Xuan, Y.A.; Chen, G. An artificial neural network for the residual isobaric heat capacity of liquid HFC and HFO refrigerants. Int. J. Refrig. 2019, 98, 381–387. [Google Scholar] [CrossRef]
- Gharagheizi, F.; Eslamimanesh, A.; Mohammadi, A.H.; Richon, D. Determination of Critical Properties and Acentric Factors of Pure Compounds Using the Artificial Neural Network Group Contribution Algorithm. J. Chem. Eng. Data 2011, 56, 2460–2476. [Google Scholar] [CrossRef]
- Kohberger, R. Similarity and Clustering in Chemical Information Systerns. Technometrics 1990, 32, 359–360. [Google Scholar] [CrossRef]
- Domenico, A.; Daniela, T.; Kamel, M.; Felice, M.G.; Orazio, N. Prediction of Acute Oral Systemic Toxicity Using a Multifingerprint Similarity Approach. Toxicol. Sci. 2019, 167, 484–495. [Google Scholar]
- Yi, D.A.; Mc, B.; Chao, G.A.; Peng, Z.C.; Jw, A. Molecular fingerprint-based machine learning assisted QSAR model development for prediction of ionic liquid properties. J. Mol. Liq. 2021, 326, 115212. [Google Scholar]
- Bahadori, A.; Tavalaeian, M.; Soleimani, R.; Lee, M.; Hashemkhani, M. Prediction of the binary surface tension of mixtures containing ionic liquids using Support Vector Machine algorithms. J. Mol. Liq. 2015, 211, 534–552. [Google Scholar]
- Huo, Y.; Bouffard, F.; Joós, G. Decision tree-based optimization for flexibility management for sustainable energy microgrids. Appl. Energy 2021, 290, 116772. [Google Scholar] [CrossRef]
- Liaw, A.; Wiener, M. Classification and Regression by randomForest. R News 2002, 2, 18–22. [Google Scholar]
- Cheshmberah, F.; Fathizad, H.; Parad, G.A.; Shojaei, S. Comparison of RBF and MLP neural network performance and regression analysis to estimate carbon sequestration. Int. J. Environ. Sci. Technol. 2020, 17, 3891–3900. [Google Scholar] [CrossRef]
- Calm, J.M.; Hourahan, G.C. Physical, safety, and environmental data for current and alternative refrigerants. In Proceedings of the 23rd International Congress of Refrigeration (ICR2011), Prague, Czech Republic, 21–26 August 2011. [Google Scholar]
- Dong, J.; Cao, D.-S.; Miao, H.-Y.; Liu, S.; Deng, B.-C.; Yun, Y.-H.; Wang, N.-N.; Lu, A.-P.; Zeng, W.-B.; Chen, A.F. ChemDes: An integrated web-based platform for molecular descriptor and fingerprint computation. J. Cheminform. 2015, 7, 60. [Google Scholar]
- Schultz, H.P.; Schultz, T.P.; Schultz, E.B. Topological Organic Chemistry. 9. Graph Theory and Molecular Topological Indices of Stereoisomeric Organic Compounds. J. Chem. Inf. Comput. Sci. 1995, 35, 864–870. [Google Scholar] [CrossRef]
- Haruo, H. Topological Index. A Newly Proposed Quantity Characterizing the Topological Nature of Structural Isomers of Saturated Hydrocarbons. Bull. Chem. Soc. Jpn. 1971, 44, 2332–2339. [Google Scholar]
- Steinbeck, C.; Han, Y.; Kuhn, S.; Horlacher, O.; Willighagen, E. The Chemistry Development Kit (CDK): An Open-Source Java Library for Chemo- and Bioinformatics. ChemInform 2003, 34, 493–500. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Fingerprints | MACCS | Pubchem | Extended | Morgan |
|---|---|---|---|---|
| Length | 166 | 881 | 1024 | 2048 |
| After removal | 42 | 80 | 191 | 376 |
| Compounds | S | Texp/K | Tcal1/K | Deviation/% | Tcal2/K | Deviation/% |
|---|---|---|---|---|---|---|
| (Z)-1,2-Dichloroethylene | 3846 | 507.25 | 518.97 | 2.3105 | 558.45 | 10.094 |
| (E)-1,2-Dichloroethylene | 2838 | 535.8 | 533.2 | 0.4853 | 558.45 | 4.228 |
| (Z)-1,2,3,3,3-Pentafluoropropene | 7758 | 379.25 | 376.13 | 0.822 | 435.30 | 2.003 |
| (E)-1,2,3,3,3-Pentafluoropropene | 6636 | 386.75 | 376.21 | 2.727 | 435.30 | 13.789 |
| (Z)-2-Butylene | 180 | 435.5 | 437.40 | 0.436 | 430.03 | 1.257 |
| (E)-2-Butylene | 68 | 428.6 | 426.33 | 0.530 | 430.03 | 0.333 |
| 1,1,1,2,2,3-Hexafluoropropane | 8276 | 403.35 | 411.48 | 2.017 | 404.06 | 0.175 |
| 1,1,1,2,3,3-Hexafluoropropane | 8741 | 412.45 | 411.01 | 0.349 | 494.52 | 19.897 |
| 1,1,1,3,3,3-Hexafluoropropane | 8984 | 398.1 | 410.77 | 3.183 | 386.51 | 2.912 |
| 2,2,3-Trimethylpentane | 424 | 563.5 | 573.40 | 1.757 | 566.24 | 2.736 |
| 2,2,4-Trimethylpentane | 460 | 543.8 | 545.11 | 0.241 | 545.16 | 0.250 |
| 2,3,3-Trimethylpentane | 412 | 573.5 | 573.06 | 0.077 | 594.42 | 3.648 |
| 2,3,4-Trimethylpentane | 426 | 566.4 | 567.14 | 0.130 | 588.60 | 3.920 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Que, Y.; Ren, S.; Hu, Z.; Ren, J. Machine Learning Prediction of Critical Temperature of Organic Refrigerants by Molecular Topology. Processes 2022, 10, 577. https://doi.org/10.3390/pr10030577
Que Y, Ren S, Hu Z, Ren J. Machine Learning Prediction of Critical Temperature of Organic Refrigerants by Molecular Topology. Processes. 2022; 10(3):577. https://doi.org/10.3390/pr10030577
Chicago/Turabian StyleQue, Yi, Song Ren, Zhiming Hu, and Jiahui Ren. 2022. "Machine Learning Prediction of Critical Temperature of Organic Refrigerants by Molecular Topology" Processes 10, no. 3: 577. https://doi.org/10.3390/pr10030577