1. Introduction
Glioblastoma multiforme is a cancer with extremely high aggressiveness, malignancy and mortality [
1]. It is characterized by an insufficient number of therapeutic possibilities and a poor prognosis. This is due to its unrestrained proliferation, infiltrative and rapid growth, neurodegeneration and diffuse tissue penetration [
2]. This type of cancer in adults most often develops in the temporal lobe of the brain and might be composed of several types of different cells. Glioblastoma multiforme accounts for 17% of primary brain tumors. It most often develops between the ages of 50 and 70. The median survival of patients is only 14.7 months after diagnosis despite the aggressive and multifaceted therapeutic approaches that are currently being used [
3,
4]. Its worldwide incidence is 0.59–3.69 per 100,000 births and only 3% of patients survive for five years after the diagnosis [
5]. Despite the numerous studies that have been conducted over many years, the exact factors that might indicate the basis of this tumor are not known.
Because there are also no effective methods of therapy, new substances that will provide an effective cure are constantly being sought. One of these is celastrol, which is a natural, pentacyclic, triterpene quinonomethyl compound. This bioactive substance is abundant in the plant
Tripterygium wilfordii, which belongs to the Celastraceae family.
Tripterygium wilfordii is a perennial grapevine that grows in south China. It is also known as Lei Gong Teng or Thunder God Vine in various parts of the world [
6,
7]. Many preclinical studies have indicated that this compound, which has been used for centuries in Chinese alternative medicine, has an anti-cancer effect [
8]. Experience has shown that celastrol can induce autophagy, inhibit the cell cycle or the apoptosis of cancer cells [
6]. In addition, it has been proven that it is able to suppress invasiveness, inhibit the growth of the tumor itself, and, additionally, has an anti-angiogenic effect [
9]. Its influence on such key properties of tumors has prompted scientists to take a closer look at this natural substance.
Because of the poor prognosis features of glioblastoma multiforme, new methods are being sought that will effectively cure it. For this purpose, it is worth looking at microRNAs, which are small, non-coding, endogenous RNA molecules. Their role is to control gene expression by inhibiting the translation process or by binding to mRNA. MicroRNAs participate in most cellular processes such as the proliferation, differentiation, cell death and regulation of the cell cycle itself [
10,
11]. Moreover, each microRNA is able to influence mRNA either by acting as a potent oncogene or tumor suppressor [
12]. In the presented experiment, miR-9-2, miR-17 and miR-19 were used because they are overexpressed in the studied tumor [
11].
The
miR-9-2 gene is expressed almost exclusively in the brain and is involved in the development of the nervous system. It is also involved in the regulation of many neoplastic processes. Its abnormal expression has been demonstrated in various neoplasms, while also pointing to its dual role, for example, as a pro-metastasis onco-miR in breast cancer, and as a tumor suppressor in melanoma. MiR-9-2 has been shown to significantly stimulate the proliferation, migration and invasion of glioblastoma cells and promote the production of new blood vessels in vitro and in vivo. MiR-9-2 has also been implicated in angiogenesis. It has been suggested that miR-9 is an inherent onco-miR in human glioma. However, the mechanisms that underlie the miR-9-2 overexpression, functional changes, the functions of miR-9-2 in glioma angiogenesis and the molecular mechanism by which miR-9-2 influences the malignant phenotypes in glioblastoma have not yet been fully elucidated [
13].
Transcriptome analysis in glioblastoma cells showed that miR-17 expression is increased in high-grade tumors. Research results indicate that miR-17 acts specifically in the G1/S phase of the cell cycle and targets multiple genes involved in the transition between these phases. The contribution of miR-17 to cancer development is still under discussion. There are reports of both its oncogenic and suppressive role. MiR-17 is involved in regulating the autophagy process in glioblastoma. It has been shown that the inhibition of miR-17 expression activates autophagy and conditions the sensitization of GBM cells to treatment with chemotherapeutic agents and ionizing radiation [
14].
The miR-17-92 cluster, to which miR-19 belongs, is considered the first miRNA cluster with oncogenic potential. It contains 6 single mature miRNAs, and miR-19 is a key oncogenic miRNA among the six members of miR-17-92 cluster. It has been shown that the expression of the miR-17-92 cluster is increased in glioblastoma multiforme. MiR-19 expression is increased in glioblastoma. MiR-19, a member of the miR-17-92 cluster, has been shown to play an oncogenic role in cancer formation. It is also known that miR-19 plays an important role in the pathogenesis of glioblastoma. In turn, inhibition of miR-17-92 clusters reduces cell proliferation and induces apoptosis in glioblastoma spheroid culture by increasing the expression of CDKN1A (cyclin-dependent kinase inhibitor 1A), E2F1, and PTEN. The participation of miR-19 in the glioblastoma process was confirmed as a recurrence.
In addition, miR-19 plays an important role in glioblastoma progression. MiR-19 is also considered a prognostic biomarker for glioma. It has been shown that high miR-19 expression in the patient’s serum is associated with poor survival. MiR-19 influences many biological characteristics of cancer cells by regulating target genes. The effect of miR-19 on the proliferation, apoptosis and migration of glioblastoma cells, and the effect of miR-19 on chemotherapy and radiotherapy have been demonstrated. Research results indicate that miR-19 inhibits the apoptosis of glioblastoma cells. It has also been reported that miR-19 promotes glioblastoma progression by the direct suppression of PPARα (peroxisome proliferator-activated α receptor, PPARα). It has also been reported that miR-19 promotes invasion and migration of glioblastoma cells by direct suppression of RhoB [
15]. Therefore,
miR-9-2,
miR-19 and
miR-17 can be identified as a potential target gene for glioblastoma therapy.
Currently, one of the molecular goals is to study the checkpoint genes that help to maintain the normal course of the cell cycle. It is believed that the genetic mutations and deletions of the cell cycle regulators could be one of the causes of this cancer [
13]. Disruption of the proper functioning of these regulators causes the escape of glioblastoma cells from the points that control the cell cycle, which will help to increase the process of their proliferation and enable a tumor to survive [
14].
Thus, the aim of our study was concerned with the changes in the cell-cycle, the proliferation and induction of apoptosis in U251MG glioblastoma multiforme cells after the miR-9-2, miR-17 and miR-19 gene knockdowns and their exposure to celastrol. The presented study was also undertaken to examine the effect of the siRNAs targeting indicated miR genes on U251MG cells susceptibility on celastrol.
4. Discussion
The resistance of cancer cells to chemotherapy is one of the greatest problems of modern clinical oncology. Glioblastoma multiforme (GBM) is one of the brain tumors for which there are currently no effective therapies. The median survival of patients with this type of cancer is 14.6 months, the median progression-free survival is 6.9 months and the five-year survival is 9.8% [
15,
16]. Because of the resistance of glioblastoma to conventional chemotherapy and radiotherapy, it is necessary to develop new, more effective methods that would use the molecular disorders characteristic of this cancer. miRNAs, which can regulate the expression of more than 60% of human genes, are of great importance in neoplastic transformation, including oncogenes, suppressor genes and the genes related to chemoresistance.
The possibility of regulating the expression of these genes based on the phenomenon of RNA interference, their expression vectors and their combination with substances of natural origin such as, for example, celastrol, could be a potential method for reducing the invasiveness of glioblastoma and additionally increasing its sensitivity to the chemotherapeutic agents used.
The use of the technique based on the phenomenon of RNA interference might turn out to be important, especially in the treatment of neoplasms of the nervous system because of the possibility of their crossing the blood–brain barrier, which impermeable to most cytostatic drugs. Because of their biochemical properties, RNA molecules are a promising therapeutic tool.
One of the newest strategies for the treatment of cancer, including brain tumors, is the use of molecules that affect the activity of miRNAs. Out of the 2.5 thousand miRNAs found in human cells, about 70% are expressed in the central nervous system, e.g., the expression of miR-9, miR-124a, miR-124b and miR-135 has only been demonstrated in nerve cells and miR-23 only in astrocytes [
17]. It is known that 256 miRNAs are overexpressed and that 95 miRNAs have a decreased expression in glioblastoma. It has been shown,
inter alia, an increased expression of miR-21, miR-10b, miR-15b, miR-16, miR-25, miR-92b, miR-93, miR-106, miR-155, miR-210, miR-17-5p, miR- 106, miR-148a and miR-196b and a decreased expression of miR132, miR-218, miR-124, miR-128a, miR-323, miR-128, miR-7, miR-181b, miR-221, miR-222, miR-31, miR-138, miR-181 and miR-379 [
18,
19]. Malkorn et al. (2010) identified 12 miRNAs: miR-9, miR-15a, miR-16, miR-17, miR-19a, miR-20a, miR-21, miR-25, miR-28, miR-130b, miR-140 and miR-210, which were overexpressed and two, miR-184 and miR-328, whose expression was underexpressed during tumor progression [
20]. Hence, the inhibition of miRNA expression in glioblastoma cells could be an interesting therapeutic option. However, despite numerous studies that have confirmed the role of miRNAs in treating this type of cancer, studies that have shown its showing therapeutic efficacy are scarce. This type of procedure may also be effective in combination with other therapeutic methods because there is evidence that the expression of specific miRNAs is important for the sensitivity of glioblastoma cells to chemo- and radiotherapy [
21], which confirms the validity of the attempts to use them in strategies for inhibiting the multidrug resistance of tumor cells [
22,
23,
24].
This study included a preliminary analysis of the effect of celastrol in combination with the silencing of the genes encoding miR-9-2, miR-17, and miR-19, which are overexpressed in glioblastoma multiforme, on the cell cycle, the expression of selected genes related to its regulation and the expression of selected genes related to the regulation of programmed cell death—apoptosis and autophagy.
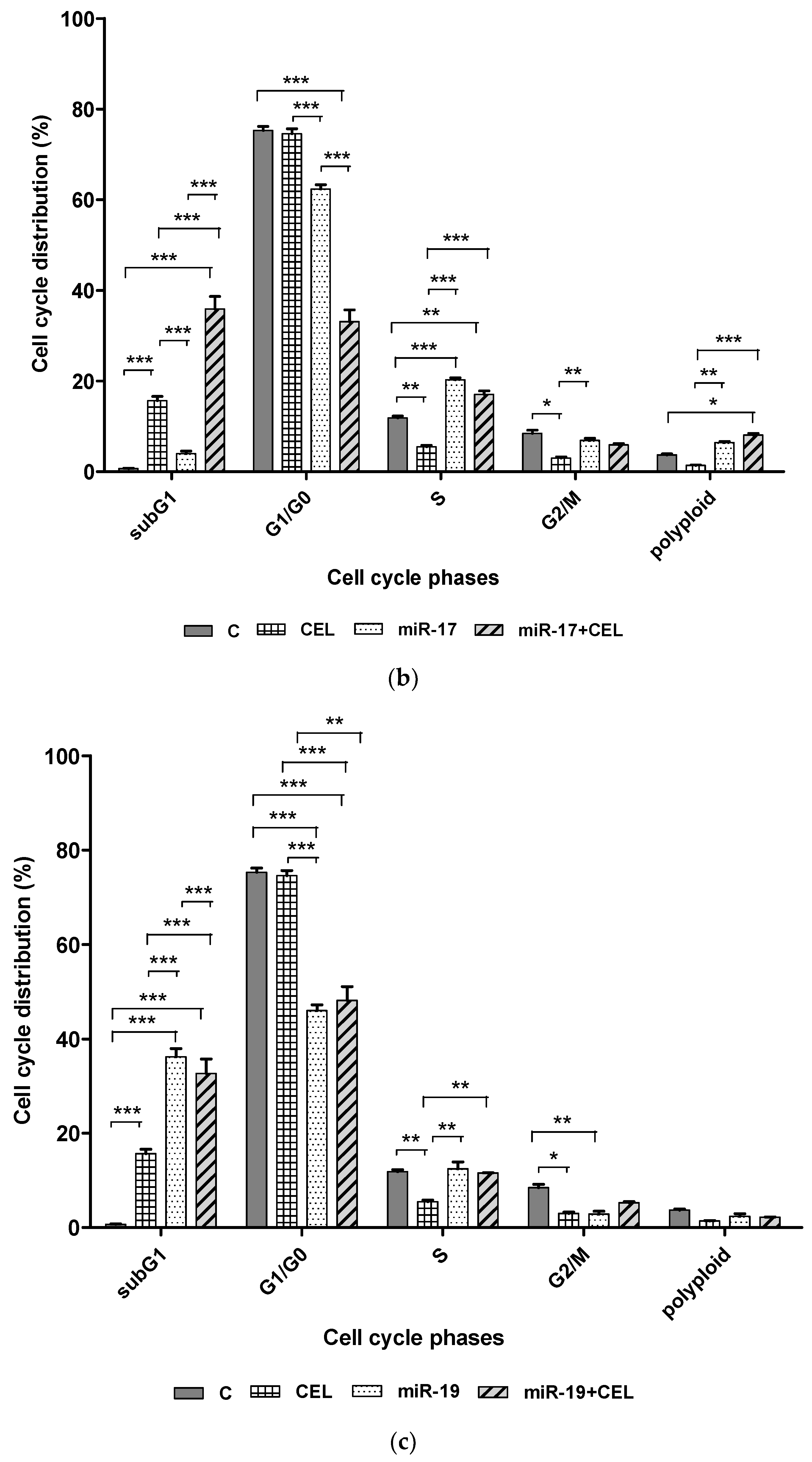
Whether silencing the genes encoding miR-9-2, miR-17, miR-19 and the exposure of U251MG glioblastoma cells to celastrol changed the percentage of cells at specific stages of the cell cycle was investigated. It was found that silencing
miR-9-2,
miR-17 and
miR-19 in combination with exposure to celastrol significantly increased the percentage of the cells of the subG1 (potentially apoptotic) population, which might suggest the induction of apoptosis and also decreased the percentage of G1/G0 cells. A similar, though slightly weaker effect was observed with the cells that had been transfected but not exposed to celastrol. The exposure of the U251MG cell line to celastrol alone was associated with a less pronounced increase in the percentage of cells in the subG1 population. It was also shown that silencing the
miR-17 and
miR-19 genes and the combination of silencing these genes with the action of celastrol and the action of celastrol alone was associated with a decrease in the percentage of the G2/M population cells. In the case of silencing the
miR-19 gene and exposure to celastrol, this was a synergistic effect. Cell cycle alteration is one of the triggers for the malignant behavior of cells seen in cancer, such as proliferation, invasion, and chemoresistance. Unfortunately, the mechanism of cell cycle regulation mediated by miR-17 is still unclear, especially in glioblastoma multiforme. In turn, the results obtained by Huang et al. (2021) indicate that miR-17 promotes cell cycle progression of the head and neck squamous cell carcinoma mainly by increasing the proportion of cells in the G2/M phase and reducing the proportion of cells in the S phase [
25]. The reduction in the percentage of cells in the G2/S phase observed by us and an increase in the percentage of cells in the S phase of the cell cycle after silencing the
miR-17 gene may indicate a similar effect on the progression of the cell cycle of glioblastoma multiforme cells.
The results of studies by other researchers indicate different mechanisms by which miR-17 promotes cell cycle progression. Cloonan et al. (2008) showed that miR-17 acts specifically at the G1/S phase boundary of the cell cycle, regulating the expression of more than 20 genes involved in the transition between these phases [
26]. In turn, Li et al. (2015) observed that miR-17 promotes the proliferation of ovarian cancer cells by promoting G1/S cell cycle transition and inhibiting apoptosis. At the same time, inhibition of
miR-17 expression was associated with the opposite effect [
27]. Contrary to our research results, Zhu et al. (2018) showed that inhibition of
miR-17 expression in pancreatic cancer cells resulted in a higher proportion of cells in the G1 phase and a lower proportion in the S phase, leading to impaired proliferation of neoplastic cells [
28]. The results obtained by various researchers indicate that miR-17 acts mainly at the G1/S boundary in promoting cell cycle progression, which was also confirmed in our research. Our results, although different from those of other researchers, may help explain the role of miR-17 in promoting cell cycle progression in glioblastoma multiforme. The observed difference may be largely due to the role of miR-17 target genes in the regulation of the cell cycle of glioblastoma.
Celastrol also caused a significant decrease in the percentage of cells in the S phase of the cell cycle, which could indicate inhibition of replication. On the other hand, it was observed that silencing the miR-17 and miR-19 genes and combining miR-17 gene silencing with the exposure of the U251MG cells to celastrol induced cell cycle arrest in S phase, which was manifested by an increased percentage of cells in this population.
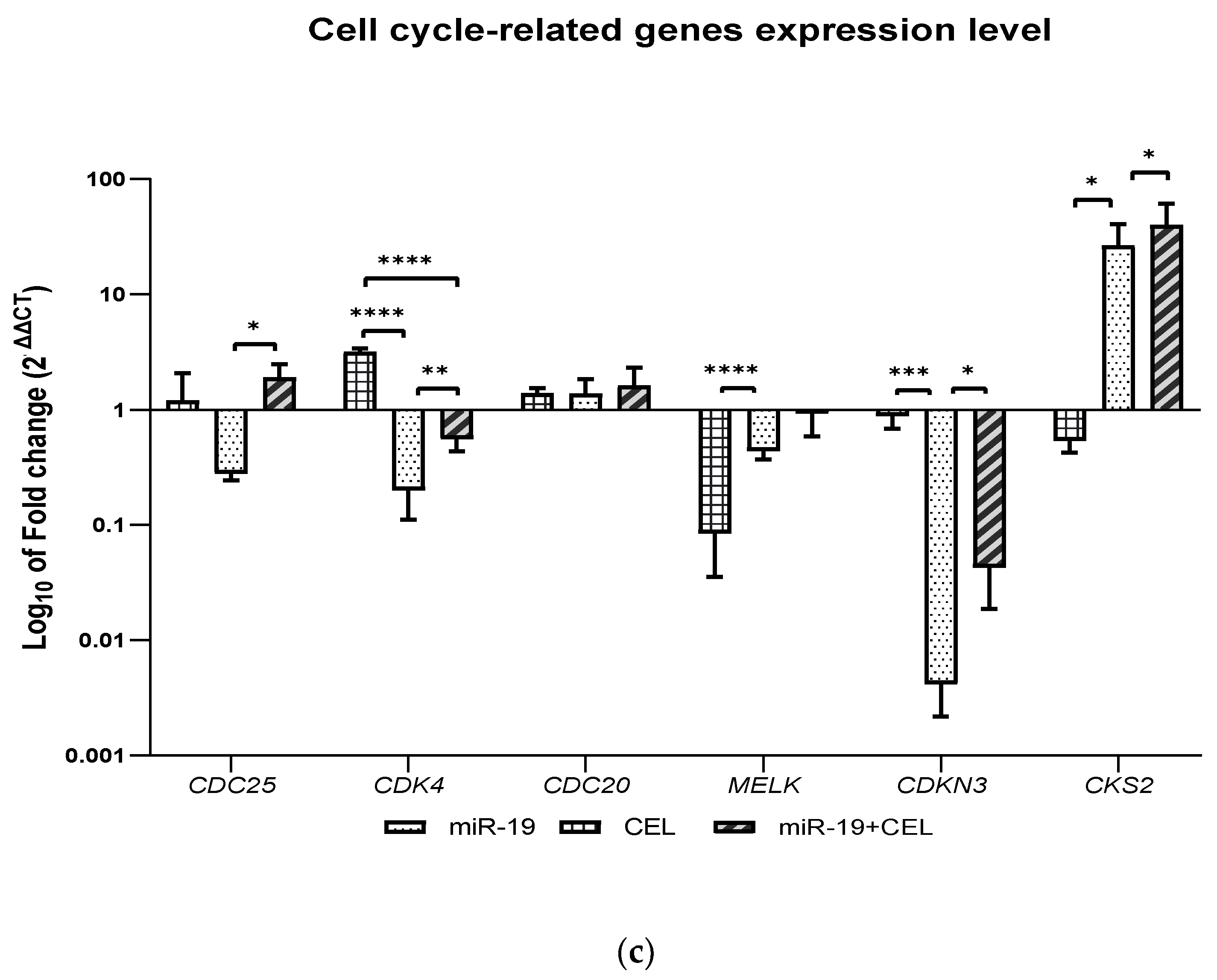
How silencing the miR-9-2, miR-17 and miR-17 genes and the exposure of glioblastoma multiforme cells of the U251MG cell line affect the expression of selected genes (CDC25A, CDK4, CDC20, MELK, CDKN3 and CKS2), that are related to the regulation of the cell cycle was also analyzed. It was shown that miR-17 and miR-19 gene silencing and the combination of miR-9-2, miR-17 and miR-19 gene silencing and exposure to celastrol decreased the expression of the CDC25A and CDK4 genes with the CDC25A expression being most strongly inhibited in the case of the silencing of the miR-9-2 gene and the exposure of tested cells to celastrol, while the silencing of the miR-19 gene and the exposure of glioblastoma multiforme U251MG cells to celastrol was associated with an increase in the CDC25A gene expression. A similar effect was observed with the exposure of the untransfected cells to celastrol.
In turn, silencing the
miR-19 gene most strongly decreased the expression of the
CDK4 gene. The CDC25 dual phosphatase family has three members: CDC25A, CDC25B and CDC25C. An overexpression of CDC25A has been documented in multiple cancer cell lines and is highly associated with malignancy and poor prognosis in cancer patients. Because CDC25A plays a more extensive role in assisting both the G1/S and G2/M progression by dephosphorylating CDK4 and CDK6, as well as CDK2 and CDK1, inhibition of the
CDC25A gene expression may cause the inhibition of the cell transition from the G1 to S phase of the cell cycle (this is possible because it is also known that the overexpression of CDC25A can accelerate the transition of cells from the G1 to S phase by prematurely increasing the activity of CDK2 kinase [
29] as well as from the G2 to M phase. The observed relationship between the miR-9, miR-17 and miR-19 gene silencing without and in combination with celastrol exposure and a decrease in the CDC25A expression and cell cycle progression requires more elucidation because that regulation of the CDC25A expression can occur at the transcriptional, translational and post-translational levels and by regulating the catalytic efficiency of CDC25A and the enzyme-substrate interaction. It should also be noted that cytoplasmic CDC25A inhibits the activity of ASK1 and increases the resistance to the apoptosis that is induced by oxidative stress and is required for Myc-dependent apoptosis [
30], hence, a decrease in the
CDC25A gene expression might increase the sensitivity of cells to the apoptosis that is induced by oxidative stress, but more detailed studies are needed to confirm this hypothesis. Moreover, because of the dual role of CDC25A in the regulation of the apoptosis process that is related to the subcellular distribution (CD25A acts as a suppressor of apoptosis only when it is in the cytoplasm and the accumulation of CDC25A in the nucleus leads to cell apoptosis), it is necessary to conduct detailed studies to explain the effect of a decrease in the CDC25A gene expression for regulating apoptosis. Since CDC25A is involved in several different biological processes, including cell division, cell proliferation, the cellular response to UV, DNA replication, cell transition from G1 phase to S phase, cell cycle regulation and regulating the activity of cyclin-dependent serine-threonine kinases, a decrease in the expression of the gene encoding this cyclin might have a multidirectional effect; therefore, it is impossible to clearly define the effects of this phenomenon.
Knockdown of the
miR-17 gene and the combination of silencing the said gene with The knockdown of the
miR-17 gene and the combination of the silencing of said gene with the exposure to celastrol decreased the expression of the C
DC20 gene, while silencing the
miR-9-2 gene and
miR-19 genes combined with the exposure of the glioblastoma multiforme cells to celastrol or the action of celastrol alone increased the expression of this gene. The results of studies by Wang et al. (2017) indicate that overexpression of CDC20 is involved in temozolomide-resistant glioma cells with an epithelial-mesenchymal transition [
31]. Decreasing the expression of the
CDC20 gene in the case of
miR-17 gene silencing might, therefore, be associated with increased sensitivity of glioblastoma cells to temozolomide; however, additional studies using other glioblastoma cell lines are necessary to confirm this hypothesis.
A decrease in the
MELK gene expression was observed after silencing the
miR-9-2 gene and exposing cells to celastrol, as well as after silencing the
miR-17 and
miR-19 genes and in the case of celastrol alone. Silencing the
miR-9-2 gene most strongly decreased the expression of the
MELK gene. The Maternal Embryonic Leucine Zipper Kinase (MELK) is overexpressed in multiple cancer types, including melanoma, colorectal cancer and triple-negative breast cancer and is a putative drug target [
32,
33,
34]. A high expression of MELK is associated with a poor patient prognosis. It was found that the knockdown of MELK using RNA interference was associated with blocking cancer cell proliferation and triggering cell cycle arrest or causing a mitotic catastrophe [
35]. The knockdown of the
miR-9-2,
miR-17 or
miR-19 gene with or without exposure to celastrol, which is associated with a significant decrease in the expression of the
MELK gene may thus result in the inhibition of the proliferation of glioblastoma multiforme cells and the induction of a mitotic catastrophe, which could be confirmed by demonstrating, for example, the presence of characteristic features within the cell nucleus and the presence of abnormal mitotic figures. Additionally, MELK has been implicated in several other cancer-related processes, for example, cancer stem cell maintenance and chemotherapy resistance; hence, the inhibition of the expression of this gene may be associated with increased sensitivity of glioblastoma cells to chemotherapy. However, more research is needed to confirm this hypothesis. We assume that additional studies will be conducted to confirm that the inhibition of the MELK gene expression could increase the sensitivity of glioblastoma cells to temozlolomide.
The knockdown of the
miR-9-2,
miR-17 and
miR-19 genes and the combination of
miR-19 gene silencing with exposure to celastrol and the action of celastrol alone decreased the expression of the
CDKN3 gene. Silencing the
miR-17 gene most strongly decreased the expression of this gene. Cyclin-dependent kinase inhibitor 3 (
CDKN3) encodes a protein from the family of protein phosphatases that has a dual specificity. CDKN3 kinase dephosphorylates CDK2 at the Thr160 position and thus prevents its activation [
36]. A CDKN3 overexpression causes the inhibition of the cell transition from the G1 to S phase [
37]. A CDKN3 overexpression is found in many human tumor tissues and tumor cell lines (hepatocellular carcinoma, lung adenocarcinoma, breast cancer, cervical cancer, ovarian cancer, gastric cancer and kidney cancer) [
38,
39,
40,
41,
42,
43]. In most cases, CDKN3 acts as an oncogene. It has been shown that, in glioblastoma cells, CDKN3 can inhibit cell proliferation and migration via a phosphatase-dependent inhibition of CDC2. Because CDKN3 can perform different biological functions in different types of cancer cells, the relationship between the silencing of the
miR-9-2,
miR-17 and
miR-19 genes and exposure to celastrol and the decrease in the
CDKN3 gene expression that was observed in this study requires a more detailed analysis to determine its importance for the survival and proliferation activity of glioblastoma cells.
We also revealed that the knockdown of the
miR-17 and
miR-19 genes and the combination of the silencing of the above-mentioned genes and the silencing of the
miR-9-2 gene combined with the exposure of glioblastoma multiforme cells to celastrol increased the expression of the
CKS2 gene, while the action of celastrol alone or the silencing of the
miR-9-2 gene decreased the expression of this gene. The
CKS2 gene expression was most strongly increased after the
miR-17 gene silencing and cell exposure to celastrol. CKS2 (CDC28 protein kinase regulatory subunit 2) belongs to the CKS family and plays an important role in early embryonic development, somatic cell division and meiosis. CKS2 has been shown to play an important role in regulating the cell cycle. It has been shown that the knockdown of the
CKS2 gene causes a decrease in the expression of cyclin A and cyclin B1 [
44]. A growing body of evidence also indicates that CKS2 drives the incidence and growth of cancer and is responsible for metastasis among many human malignancies. Some research results have indicated that
CKS2 mRNA is overexpressed in low-grade glioma and may be involved in the pathogenesis of gliomas [
45,
46]. Currently, CKS2 is considered to be a prognostic biomarker in low-grade glioma. However, the
CKS2 expression is differentiated in low-grade glioma and non-neoplastic tissues. Some authors have pointed to the correlation between the
CKS2 expression and the IDH1 mutation status.
CKS2 was shown to be upregulated in the IDH1-wildtype, while the genetic test results revealed a recurrent mutation in the isocitrate dehydrogenase (IDH1) gene in most GBM cells. Therefore, additional research is needed to understand the importance of increasing or decreasing the expression of the
CKS2 gene in GBM cells.
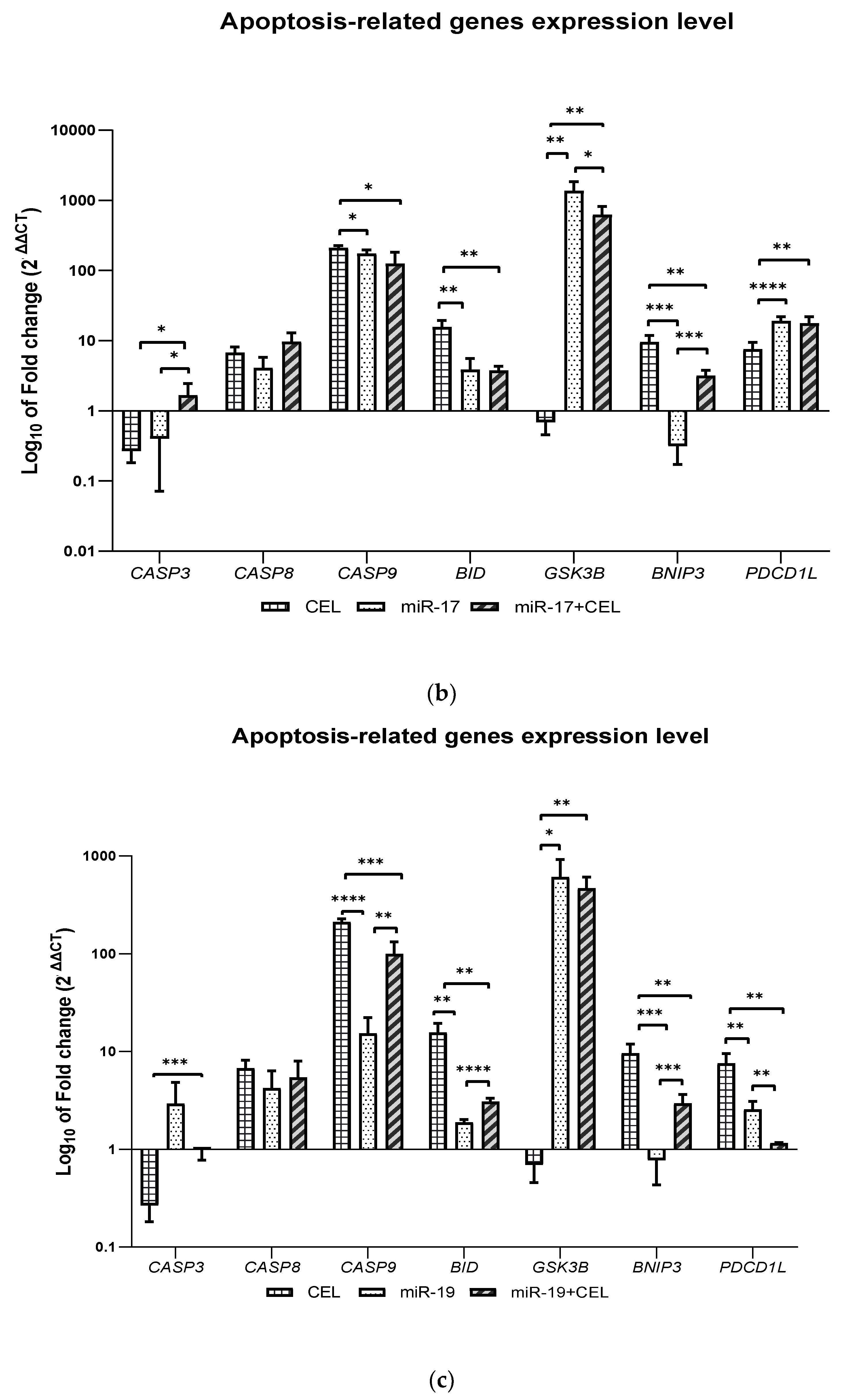
It was shown that the knockdown of the miR-9-2, miR-17 and miR-19 genes and the combination of miR-17 and miR-19 gene silencing with the exposure of glioblastoma multiforme cells to celastrol and the action of celastrol alone significantly increased the expression of the CASP8, CASP9 and BID genes, which may indicate the activation of the extrinsic and intrinsic pathways of apoptosis induction. Only in the case of the silencing of the miR-17 gene in combination with the exposure of cells to celastrol was an increase in the expression of the CASP3 gene observed. In turn, the effect of celastrol was associated with the greatest increase in the BID expression and silencing of the miR-17 gene in combination with celastrol exposure was associated with the greatest increase in the CASP8 expression, which in these cases may indicate the induction of the apoptosis of glioblastoma multiforme cells mainly via an extrinsic pathway but further studies are required to confirm this hypothesis.
However, the available data might support this hypothesis. Celastrol belongs to the group of so-called proteasome inhibitors. It is known to inhibit the CT-L activity of the 20S proteasome with an IC50 = 2.5 µM. It was shown that in the prostate cancer cells of the PC-3 and LNCaP lines, celastrol caused an increase in the level of the ubiquitinated proteins, IκBα, Bax and P27, which resulted in the induction of apoptosis. Celastrol enhances TNF- and chemotherapeutic-induced apoptosis and inhibits the proliferation and invasiveness of cancer cells by inhibiting both the inducible and constitutive activation of NF-kB [
47,
48]. One of the main goals in cancer therapy is to completely reduce the tumor cell mass, e.g., via the induction of apoptosis (type I programmed cell death). Since glioblastomas are resistant to therapies that induce apoptosis [
49], the observed phenomenon is highly desirable.
It was also observed that the
GSK3B gene expression was decreased only when
miR-9-2 gene silencing was combined with the exposure of glioblastoma cells to celastrol. In the remaining cases, an increase in the expression of this gene was observed and the observed effect was the strongest after silencing the
miR-17 gene, which was also associated with the lowest expression of the
BNIP3 gene. In turn, the action of celastrol was associated with the greatest increase in the expression of this gene. GSK3B kinase (glycogen synthase kinase-3 beta) is involved in cellular metabolism, neuronal cell development and body pattern formation by triggering the degradation of the signaling or functional proteins. GSK3B is involved in regulating both apoptosis and autophagy. The role of GSK3B in the regulation of apoptosis is ambiguous; namely, GSK3B can act as both a pro-apoptotic and anti-apoptotic factor depending on the type of cell and the surrounding environment. GSK3β is able to promote apoptosis and DNA damage under hypoxic conditions by inhibiting the cell “survival” signals such as the cAMP response element-binding protein (CREB), heat shock protein 1 and by activating the pro-apoptotic transcription factors such as P53. However, inhibiting GSK3β synthesis could lead to the hypophosphorylation of the MDM2 protein, which in turn suggests an anti-apoptotic effect of GSK3β [
50]. The activation of PI3KCA that was observed in the case of glioblastoma multiforme induced the phosphorylation of GSK3B at Ser9 and attenuated the interaction of GSK3B with Bcl-2, which prevented the phosphorylation of Bcl-2 at Ser70 and was connected with the ubiquitin-mediated degradation of Bcl-2. In turn, the increased expression of Bcl-2 interferes with the activation of BECN1 and attenuates autophagy in cancer cells [
51]. Several studies have indicated that GBM cells seem to be less resistant to therapies that induce cell death that has the features of autophagy (type II programmed cell death) [
52]. Our previous research results also showed that the siRNAs that are specific for the
AKT3 and
PI3KCA genes decreased the
BNIP3 mRNA expression [
53]. Although BNIP3 (Bcl-2/adenovirus E1B 19 kDa interacting protein 3) is a member of the pro-cell death Bcl-2 family, its proapoptotic activity is questionable. BNIP3 is known to induce autophagy and plays a key role in As
2O
3-induced autophagic cell death in malignant glioma cells [
54]. BNIP3 is nuclear and is localized in most GBMs and does not cause cell death. It has been shown that BNIP3 binds to the promoter of the AIF gene, inhibits its expression and inhibits temozolomide-induced apoptosis (TMZ) in glioblastoma cells [
55]. Because there was a reduced copy number of
BNIP3 mRNA in the transfected GBM cells, transfected cells may therefore be associated with the induction of apoptosis. The serine-threonine kinase ULK1, which is encoded by the gene of the same name, is a key initiator of autophagy.
It has been shown that silencing the miR-9-2 gene and the combination of the knockdown of this gene with exposure to celastrol increases the expression of the BECN1 and ULK1 genes. Moreover, it was found that silencing the miR-17 and miR-19 genes in cells that had either been exposed or not exposed to celastrol not only increased the expression of the BECN1 and ULK1 genes, but also that of the MAPLC3IIA gene.
The obtained results may therefore indicate the induction of autophagy. We observed the induction of autophagy and apoptosis after the knockdown of the
miR-9-2,
miR-17 and
miR-19 genes and the combination of the silencing of these genes with the exposure to celastrol, which is consistent with the available results of other studies confirming the possibility of the simultaneous activation of these processes. However, our results are difficult to interpret unequivocally because of the dual role of autophagy—either promoting or inhibiting cancer [
56]. Autophagy can both stimulate and prevent cancer depending on the cell type and the cellular context [
57]. Additionally, the results of some studies suggest that inhibiting autophagy at different time stages may produce different results [
58,
59]. Thus, more research is needed to elucidate the exact mechanism of autophagy and the induction apoptosis in GBM cells after the knockdown of
miR-9-2,
miR-17 and
miR-19 and whether the induction of autophagy is a positive phenomenon in increasing the sensitivity of GBM cells to the chemotherapeutic agents—temozolomide and carmustine.
The significance of the presented research results is cognitive. Preliminary analysis of the effect of silencing selected genes encoding miRNAs overexpressed in glioblastoma (miR-9-2, miR-17 and miR-19) and exposure of GBM cells to celastrol was to characterize the type of changes occurring in the expression of selected genes related to the regulation of cellular processes important for the proliferation and progression of this tumor. Demonstrating the relationship between the silencing of miR-9-2, miR-17 and miR-19 and changes in the expression of these genes is the basis for determining the mechanism by which such a change occurs and what its consequences will be. The results obtained by us should make it possible to determine the validity of silencing the genes encoding the indicated miRs in combination with celastrol in the experimental therapy of glioblastoma. Evidence of the effectiveness of regulation of the expression of selected miRNA-coding genes in glioblastoma multiforme cell cultures may be the basis for confirming the obtained effects in vivo on the basis of examining the increase in tumor mass and monitoring the survival time of animals with cancerous tumors. This precedes possible clinical trials. The results of this study could therefore form the basis for the development of new and more perfect methods for therapy of glioblastoma multiforme.


 ,
, 









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}