
18p Deletion Syndrome Originating from Rare Unbalanced Whole-Arm Translocation between Chromosomes 13 and 18: A Case Report and Literature Review


Abstract
:1. Introduction
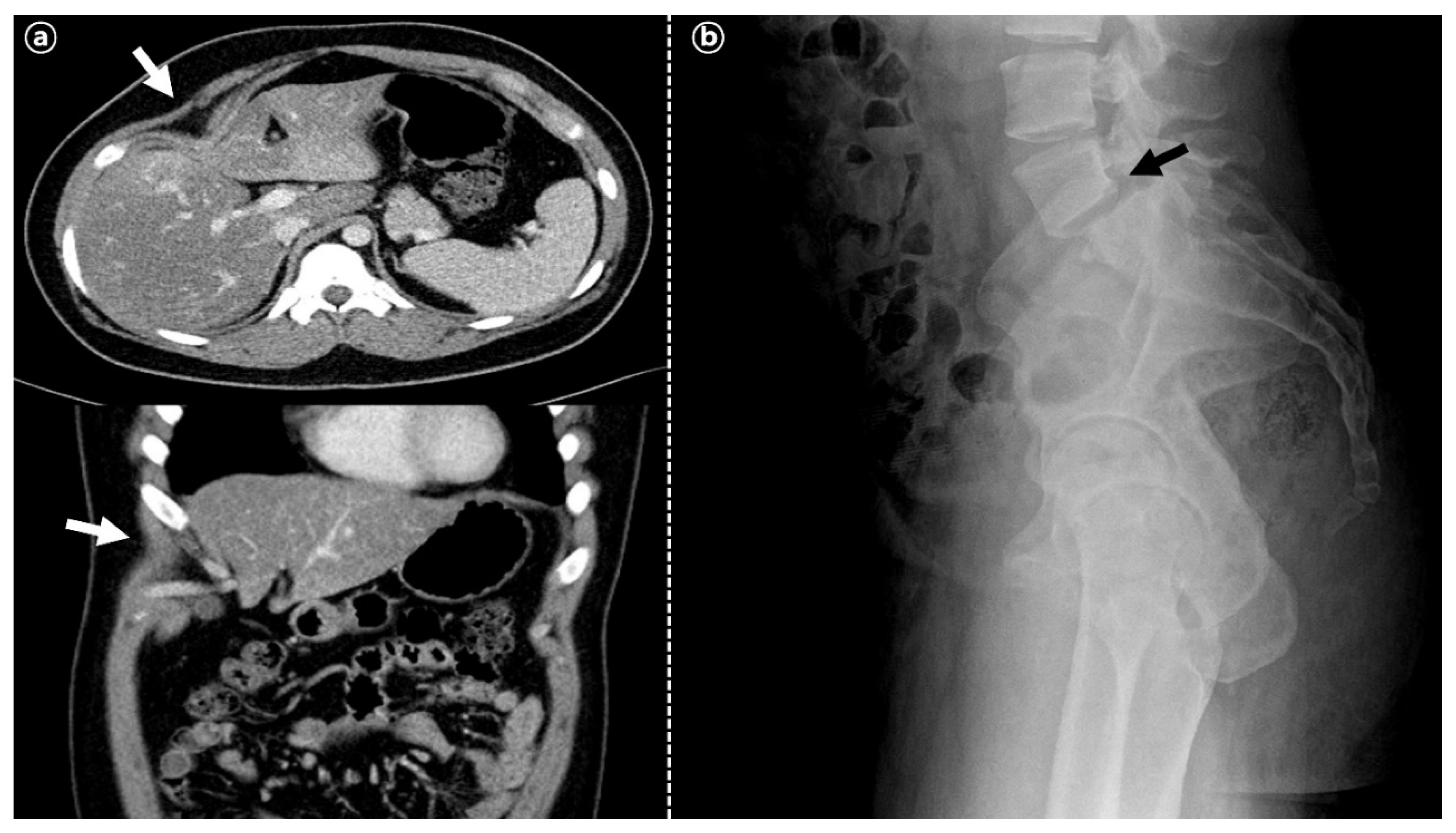
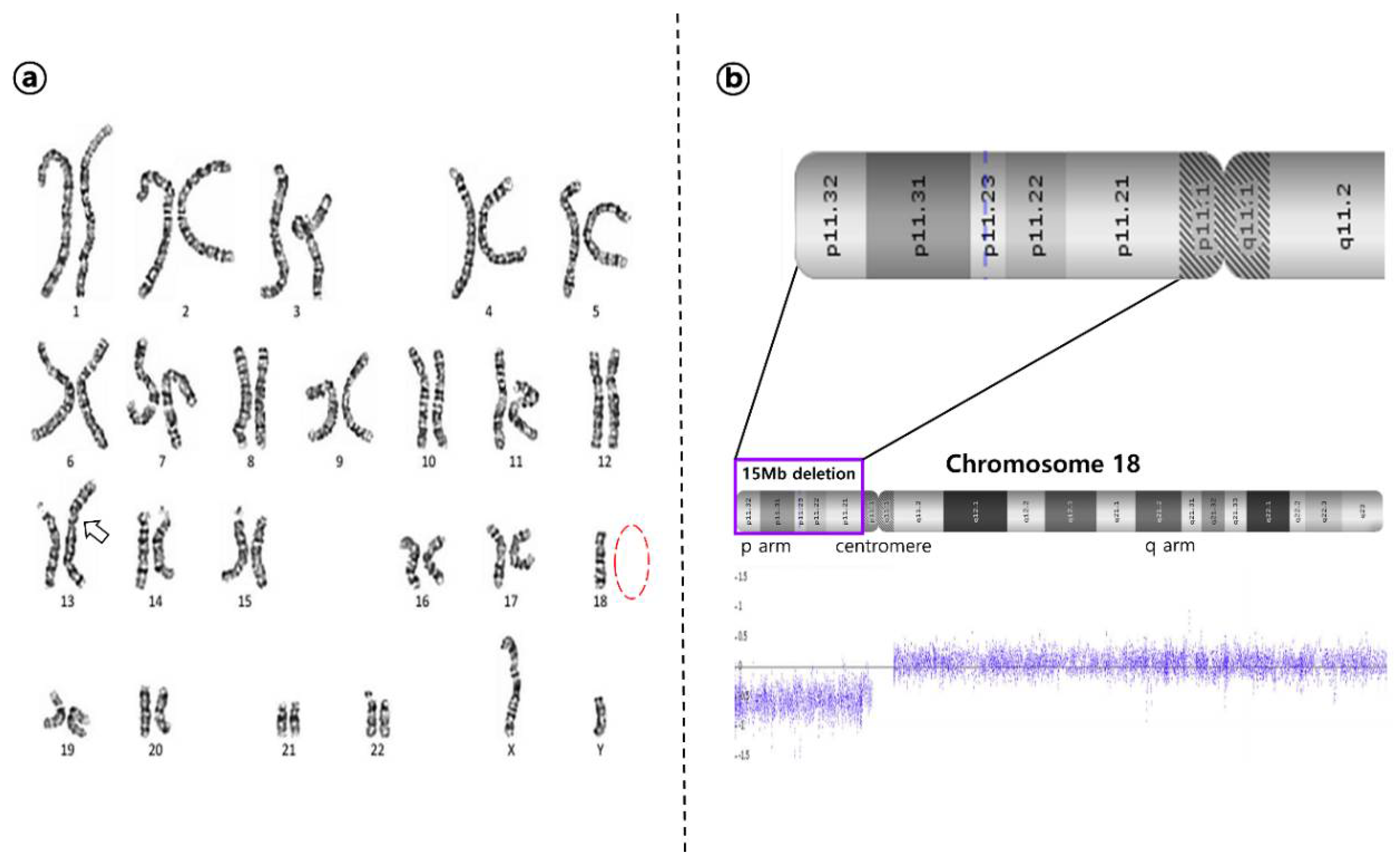
2. Case Description
3. Discussion
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Turleau, C. Monosomy 18p. Orphanet J. Rare Dis. 2008, 3, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, A.; Kim, J.; Cho, S.Y.; Lee, J.-E.; Kim, H.-J.; Jin, D.-K. A case of de novo 18p deletion syndrome with panhypopituitarism. Ann. Pediatr. Endocrinol. Metab. 2019, 24, 60–63. [Google Scholar] [CrossRef] [PubMed]
- McGhee, E.M.; Qu, Y.; Wohlferd, M.M.; Goldberg, J.D.; Norton, M.E.; Cotter, P.D. Prenatal diagnosis and characterization of an unbalanced whole arm translocation resulting in monosomy for 18p. Clin. Genet. 2001, 59, 274–278. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.-C.C.; Nemana, L.; Kou, S.Y.; Habibian, R.; Hajianpour, M.J. Molecular cytogenetic characterization of 18;21 whole arm translocation associated with monosomy 18p. Am. J. Med. Genet. 1997, 71, 463–466. [Google Scholar] [CrossRef]
- Moedjono, J.S.; Funderburk, S.J.; Sparkes, R.S. 18p--syndrome resulting from translocation (13a;18q) in a mildly affected adult male. J. Med. Genet. 1979, 16, 399–402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Ravel, T.J.; Thiry, P.; Fryns, J.P. Follow-up of adult males with chromosome 18p deletion. Eur. J. Med. Genet. 2005, 48, 189–193. [Google Scholar] [CrossRef] [PubMed]
- Nema, D.S.R.; Venkatnarayan, K.; Dalal, S.; Sodhi, K. A Rare Association of Monosomy 18 with Translocation 13p 11/18 with Cholelithiasis. J. Pediatr. Neonatal. Care 2016, 4, 00148. [Google Scholar]
- Safavi, M.; Ashtiani, M.T.H.; Badv, R.S.; Azari-Yam, A.; Vasei, M. A Rare Cytogenetic Variant of Monosomy 18p Syndrome as a Consequence of Whole-Arm Translocation between Chromosomes 13 and 18. Arch. Iran Med. 2019, 22, 627–628. [Google Scholar] [PubMed]
- Hasi-Zogaj, M.; Sebold, C.; Heard, P.; Carter, E.; Soileau, B.; Hill, A.; Rupert, D.; Perry, B.; Atkinson, S.; O’Donell, L.; et al. A review of 18p deletions. Am. J. Med. Genet. C Semin. Med. Genet. 2015, 169, 251–264. [Google Scholar] [CrossRef] [PubMed]
- Brenk, C.H.; Prott, E.-C.; Trost, D.; Hoischen, A.; Walldorf, C.; Radlwimmer, B.; Wieczorek, D.; Propping, P.; Gillessen-Kaesbach, G.; Weber, R.G.; et al. Towards mapping phenotypical traits in 18p- syndrome by array-based comparative genomic hybridisation and fluorescent in situ hybridisation. Eur. J. Hum. Genet. 2007, 15, 35–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, Q.; Qiang, R.; Cai, B.; Wang, X.; Cai, N.; Zhen, S.; Zhai, W. The genotype and phenotype of chromosome 18p deletion syndrome: Case series. Medicine 2021, 100, e25777. [Google Scholar] [CrossRef] [PubMed]
- Wester, U.; Bondeson, M.-L.; Edeby, C.; Anneren, G. Clinical and molecular characterization of individuals with 18p deletion: A genotype-phenotype correlation. Am. J. Med. Genet. A 2006, 140, 1164–1171. [Google Scholar] [CrossRef] [PubMed]
- Chiang, H.-L.; Fuh, J.-L.; Tsai, Y.-S.; Soong, B.-W.; Liao, Y.-C.; Lee, Y.-C. Expanding the phenotype of AFG3L2 mutations: Late-onset autosomal recessive spinocerebellar ataxia. J. Neurol. Sci. 2021, 428, 117600. [Google Scholar] [CrossRef] [PubMed]
- Miller, G.; Mowrey, P.N.; Hopper, K.D.; Frankel, C.A.; Ladda, R.L. Neurologic manifestations in 18q- syndrome. Am. J. Med. Genet. 1990, 37, 128–132. [Google Scholar] [CrossRef] [PubMed]
- Mello, C.B.; Bueno, O.F.A.; Benedetto, L.M.; Pimenta, L.S.E.; Takeno, S.S.; Melaragno, M.I.; Meloni, V.A. Intellectual, adaptive and behavioural characteristics in four patients with 18p deletion syndrome. J. Intellect. Disabil. Res. 2019, 63, 225–232. [Google Scholar] [CrossRef] [PubMed]
- Zuhlke, C.; Mikat, B.; Timmann, D.; Wieczorek, D.; Gillessen-Kaesbach, G.; Burk, K. Spinocerebellar ataxia 28: A novel AFG3L2 mutation in a German family with young onset, slow progression and saccadic slowing. Cerebellum Ataxias 2015, 2, 19. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
| Variables | Case 1 | Case 2 | Case 3 | Case 4 | Our Case |
|---|---|---|---|---|---|
| Age at diagnosis | 27 years | 22 years | 8 years | 1.5 years | 16 years |
| Sex | Male | Male | Female | Male | Male |
| Cytogenetic methodology | Karyotyping | Karyotyping, FISH | Karyotyping | Karyotyping | Karyotyping, aCGH |
| Karyotype | 45, XY, −13, 18, +t(13;18) (13qter→cen→18qter) | 45, XY, der(13;18) (q10;q10) | 45, XX, t(13p;18p) | 45, XY, der(13;18) (q10;q10) | 45, XY, der(13;18) (q10;q10) |
| Inheritance | de novo | de novo | NA | NA | de novo |
| Brain imaging | NA | NA | Small foci in frontal and parietal region a | Normal | Normal |
| Facial dysmorphism | Posteriorly rotated ears, short neck, dental caries, café-au-lait spot | Triangular broad face, large sloping forehead, epicanthal folds, long and broad nose, large low-set ears, short neck, alopecia, disarrayed teeth, dental caries | Dolichocephaly, hypertelorism, flat nasal bridge, high arched palate, prominent forehead | Brachycephaly, hypertelorism, protruding eyes, hypodontia | Round face, hypertelorism, flat and long nose, short neck |
| Low birth weight | (-) | (+), 1300 g | (+), 2250 g | NA | (-) |
| Short stature | (+) | (+) | (+) | (+) | (+) |
| Language disorder | (+) | (+) | (+) | (+) | (+) |
| DD/ID b | Borderline | Moderate | Mild | Global DD c | Mild |
| Behavioral features | Impulse control disorder | Intermittent explosive disorder | (-) | NA | (-) |
| Neurological features | (-) | (-) | Seizures | Seizures | Seizures, ataxic-like movement, dysarthria |
| Ophthalmologic features | Strabismus | Strabismus, ptosis, nystagmus | Strabismus, microcornea | Ptosis | Strabismus, ptosis, nystagmus, pseudopapilledema |
| Cardiac features | NA | NA | NA | Patent ductus arteriosus, pulmonary atresia, ventricular septal defect | (-) |
| Endocrinological features | (-) | Acquired hypothyroidism at 40 s | (-) | NA | (-) |
| Skeletal features | Short left 4th metacarpal bone | Mild kyphosis, pectus carinatum | (-) | NA | 8th rib cage deformity, spondylolisthesis of L5 on S1 |
| Other features | Cutaneous basal cell carcinoma (right arm) | (-) | Everted umbilicus, gall bladder calculi | (-) | Morbid obesity, micropenis |
| References (year) | Moedjono SJ et al. [5] 1979 | de Ravel TJ et al. [6] 2005 | Nema et al. [7] 2016 | Safavi et al. [8] 2019 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Choi, J.Y.; Moon, J.U.; Yoon, D.H.; Yim, J.; Kim, M.; Jung, M.H. 18p Deletion Syndrome Originating from Rare Unbalanced Whole-Arm Translocation between Chromosomes 13 and 18: A Case Report and Literature Review. Children 2022, 9, 987. https://doi.org/10.3390/children9070987
Choi JY, Moon JU, Yoon DH, Yim J, Kim M, Jung MH. 18p Deletion Syndrome Originating from Rare Unbalanced Whole-Arm Translocation between Chromosomes 13 and 18: A Case Report and Literature Review. Children. 2022; 9(7):987. https://doi.org/10.3390/children9070987
Chicago/Turabian StyleChoi, Ji Young, Ja Un Moon, Da Hye Yoon, Jisook Yim, Myungshin Kim, and Min Ho Jung. 2022. "18p Deletion Syndrome Originating from Rare Unbalanced Whole-Arm Translocation between Chromosomes 13 and 18: A Case Report and Literature Review" Children 9, no. 7: 987. https://doi.org/10.3390/children9070987