
Prematurity and Pulmonary Vein Stenosis: The Role of Parenchymal Lung Disease and Pulmonary Vascular Disease

 , and
, and 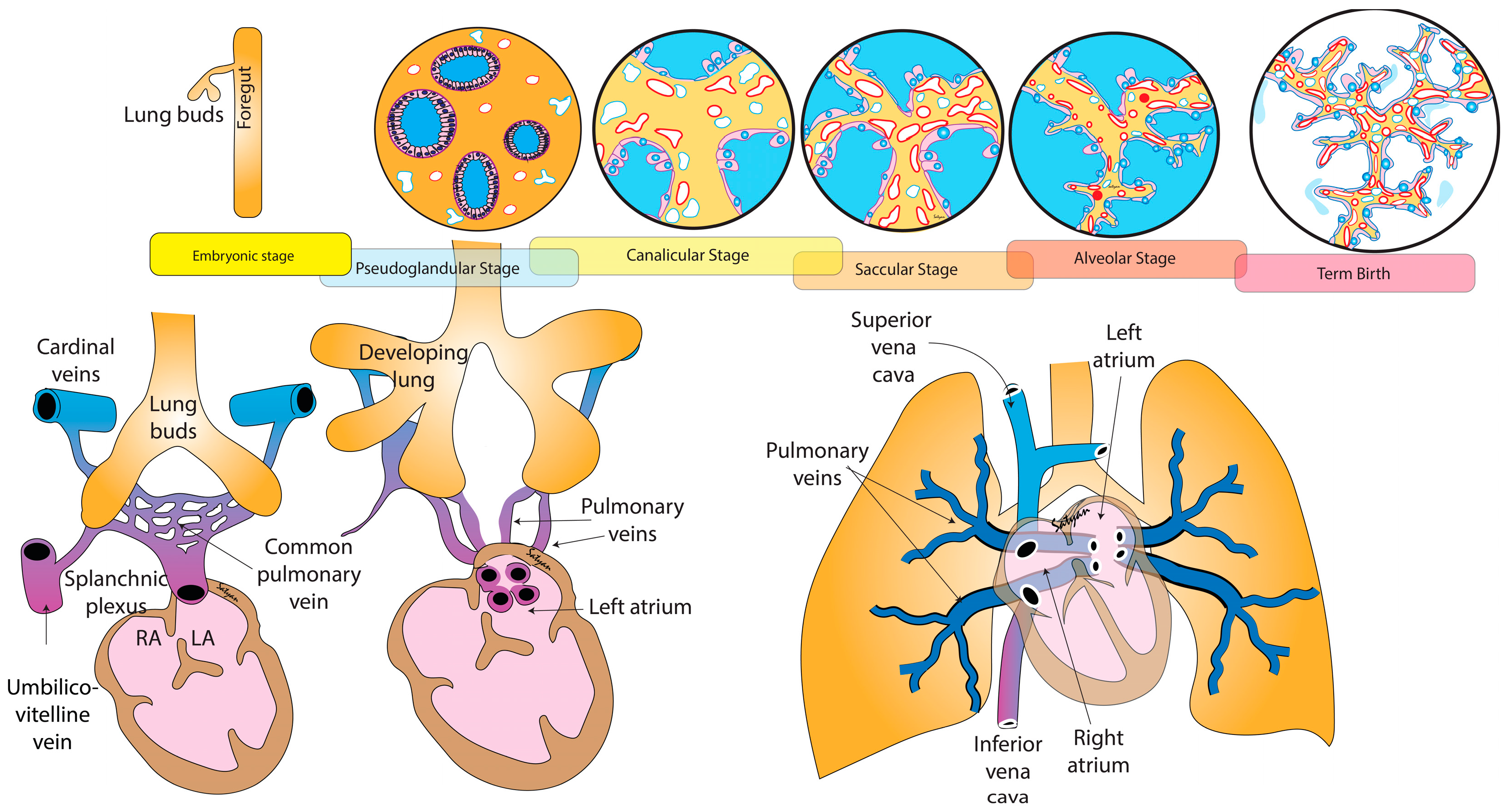{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Cardiac Morphogenesis and Airway Growth Influence Pulmonary Vascular Development
2.1. Cardiac Morphogenesis
2.2. Development of the Airways
2.3. Development of Pulmonary Vasculature
2.4. Vasculogenesis and Angiogenesis
3. Consequences of Premature Birth on PVS
3.1. Parenchymal Lung Disease Influences Vascular Development
3.2. Immature Cardiac Development
3.3. Epidemiology of PVS in Premature Infants
4. BPD Is an Inflammatory State: Relevance to PVS
5. Additional Neonatal Risk Factors
5.1. Necrotizing Enterocolitis
5.2. Left-to-Right Shunt Physiology
5.3. Fetal Growth Restriction
5.4. Genetic Linkage
5.5. Twin Pregnancy
5.6. Retinopathy of Prematurity
6. Clinical Presentation of PVS in Premature Infants
7. Challenge of PVS Detection in the NICU: Algorithm for Detection PVS in Premature Infants
8. Conclusions and Future Research
Author Contributions
Funding
Conflicts of Interest
References
- Nasr, V.G.; Callahan, R.; Wichner, Z.; Odegard, K.C.; DiNardo, J.A. Intraluminal Pulmonary Vein Stenosis in Children. Anesth. Analg. 2019, 129, 27–40. [Google Scholar] [CrossRef] [PubMed]
- Backes, C.H.; Nealon, E.; Armstrong, A.K.; Cua, C.L.; Mitchell, C.; Krishnan, U.; Vanderlaan, R.D.; Song, M.K.; Viola, N.; Smith, C.V.; et al. Pulmonary Vein Stenosis in Infants: A Systematic Review, Meta-Analysis, and Meta-Regression. J. Pediatr. 2018, 198, 36–45.e3. [Google Scholar] [CrossRef] [PubMed]
- Jadcherla, A.V.; Backes, C.H.; Cua, C.L.; Smith, C.V.; Levy, P.T.; Ball, M.K. Primary Pulmonary Vein Stenosis: A New Look at a Rare but Challenging Disease. Neoreviews 2021, 22, e296–e308. [Google Scholar] [CrossRef] [PubMed]
- Kalfa, D.; Belli, E.; Bacha, E.; Lambert, V.; di Carlo, D.; Kostolny, M.; Nosal, M.; Horer, J.; Salminen, J.; Rubay, J.; et al. Outcomes and prognostic factors for postsurgical pulmonary vein stenosis in the current era. J. Thorac. Cardiovasc. Surg. 2018, 156, 278–286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sadr, I.M.; Tan, P.E.; Kieran, M.W.; Jenkins, K.J. Mechanism of pulmonary vein stenosis in infants with normally connected veins. Am. J. Cardiol. 2000, 86, 577–579. [Google Scholar] [CrossRef]
- Frank, D.B.; Levy, P.T.; Stiver, C.A.; Boe, B.A.; Baird, C.W.; Callahan, R.M.; Smith, C.V.; Vanderlaan, R.D.; Backes, C.H. Primary pulmonary vein stenosis during infancy: State of the art review. J. Perinatol. 2021, 41, 1528–1539. [Google Scholar] [CrossRef]
- Mahgoub, L.; Kaddoura, T.; Kameny, A.R.; Lopez Ortego, P.; Vanderlaan, R.D.; Kakadekar, A.; Dicke, F.; Rebeyka, I.; Calderone, C.A.; Redington, A.; et al. Pulmonary vein stenosis of ex-premature infants with pulmonary hypertension and bronchopulmonary dysplasia, epidemiology, and survival from a multicenter cohort. Pediatr. Pulmonol. 2017, 52, 1063–1070. [Google Scholar] [CrossRef]
- Jensen, E.A.; Dysart, K.; Gantz, M.G.; McDonald, S.; Bamat, N.A.; Keszler, M.; Kirpalani, H.; Laughon, M.M.; Poindexter, B.B.; Duncan, A.F.; et al. The Diagnosis of Bronchopulmonary Dysplasia in Very Preterm Infants. An Evidence-based Approach. Am. J. Respir. Crit. Care Med. 2019, 200, 751–759. [Google Scholar] [CrossRef]
- DiLorenzo, M.P.; Santo, A.; Rome, J.J.; Zhang, H.; Faerber, J.A.; Mercer-Rosa, L.; Hopper, R.K. Pulmonary Vein Stenosis: Outcomes in Children With Congenital Heart Disease and Prematurity. Semin. Thorac. Cardiovasc. Surg. 2019, 31, 266–273. [Google Scholar] [CrossRef]
- Levy, P.T.; Jain, A.; Nawaytou, H.; Teitel, D.; Keller, R.L.; Fineman, J.; Steinhorn, R.H.; Abman, S.H.; Mcnamara, P.J.; for the Pediatric Pulmonary Hypertension Network (PPHNet). Risk Assessment and Monitoring of Chronic Pulmonary Hypertension in Premature Infants. J. Pediatr. 2019, 217, 199–209.e4. [Google Scholar] [CrossRef]
- Laux, D.; Rocchisani, M.-A.; Boudjemline, Y.; Gouton, M.; Bonnet, D.; Ovaert, C. Pulmonary Hypertension in the Preterm Infant with Chronic Lung Disease can be Caused by Pulmonary Vein Stenosis: A Must-Know Entity. Pediatr. Cardiol. 2016, 37, 313–321. [Google Scholar] [CrossRef] [PubMed]
- Heching, H.J.; Turner, M.; Farkouh-Karoleski, C.; Krishnan, U. Pulmonary vein stenosis and necrotising enterocolitis: Is there a possible link with necrotising enterocolitis? Arch. Dis. Child. Fetal Neonatal Ed. 2014, 99, F282–F285. [Google Scholar] [CrossRef] [PubMed]
- Benjamin, J.T.; Hamm, C.R.; Zayek, M.; Eyal, F.G.; Carlson, S.; Manci, E. Acquired left-sided pulmonary vein stenosis in an extremely premature infant: A new entity? J. Pediatr. 2009, 154, 459–459.e1. [Google Scholar] [CrossRef] [PubMed]
- Swier, N.L.; Richards, B.; Cua, C.L.; Lynch, S.K.; Yin, H.; Nelin, L.D.; Smith, C.V.; Backes, C.H. Pulmonary Vein Stenosis in Neonates with Severe Bronchopulmonary Dysplasia. Am. J. Perinatol. 2016, 33, 671–677. [Google Scholar] [CrossRef]
- Paige, S.L.; Plonowska, K.; Xu, A.; Wu, S.M. Molecular regulation of cardiomyocyte differentiation. Circ. Res. 2015, 116, 341–353. [Google Scholar] [CrossRef] [Green Version]
- Haddad, F.; Hunt, S.A.; Rosenthal, D.N.; Murphy, D.J. Right Ventricular Function in Cardiovascular Disease, Part I: Anatomy, Physiology, Aging, and Functional Assessment of the Right Ventricle. Circulation 2008, 117, 1436–1448. [Google Scholar] [CrossRef]
- Wu, M. Mechanisms of Trabecular Formation and Specification During Cardiogenesis. Pediatr. Cardiol. 2018, 39, 1082–1089. [Google Scholar] [CrossRef]
- Tan, C.M.J.; Lewandowski, A.J. The Transitional Heart: From Early Embryonic and Fetal Development to Neonatal Life. Fetal Diagn. Ther. 2020, 47, 373–386. [Google Scholar] [CrossRef]
- Vincent, M.; Karolak, J.A.; Deutsch, G.; Gambin, T.; Popek, E.; Isidor, B.; Szafranski, P.; Le Caignec, C.; Stankiewicz, P. Clinical, Histopathological, and Molecular Diagnostics in Lethal Lung Developmental Disorders. Am. J. Respir. Crit. Care Med. 2019, 200, 1093–1101. [Google Scholar] [CrossRef]
- Lau, E.M.; Manes, A.; Celermajer, D.S.; Galie, N. Early detection of pulmonary vascular disease in pulmonary arterial hypertension: Time to move forward. Eur. Heart J. 2011, 32, 2489–2498. [Google Scholar] [CrossRef]
- deMello, D.E.; Reid, L.M. Embryonic and early fetal development of human lung vasculature and its functional implications. Pediatr. Dev. Pathol. 2000, 3, 439–449. [Google Scholar] [CrossRef] [PubMed]
- Hislop, A.A.; Pierce, C.M. Growth of the vascular tree. Paediatr. Respir. Rev. 2000, 1, 321–327. [Google Scholar] [CrossRef] [PubMed]
- de Wijs-Meijler, D.P.; Duncker, D.J.; Tibboel, D.; Schermuly, R.T.; Weissmann, N.; Merkus, D.; Reiss, I.K.M. Oxidative injury of the pulmonary circulation in the perinatal period: Short- and long-term consequences for the human cardiopulmonary system. Pulm. Circ. 2017, 7, 55–66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malloy, K.W.; Austin, E.D. Pulmonary hypertension in the child with bronchopulmonary dysplasia. Pediatr. Pulmonol. 2021, 56, 3546–3556. [Google Scholar] [CrossRef] [PubMed]
- Gong, J.; Feng, Z.; Peterson, A.L.; Carr, J.F.; Vang, A.; Braza, J.; Choudhary, G.; Dennery, P.A.; Yao, H. Endothelial to mesenchymal transition during neonatal hyperoxia-induced pulmonary hypertension. J. Pathol. 2020, 252, 411–422. [Google Scholar] [CrossRef]
- El-Khuffash, A.; McNamara, P.J. Hemodynamic assessment and monitoring of premature infants. Clin. Perinatol. 2017, 44, 377–393. [Google Scholar] [CrossRef]
- Rayment, N.B.; Haven, A.J.; Madden, B.; Murday, A.; Trickey, R.; Shipley, M.; Davies, M.J.; Katz, D.R. Myocyte loss in chronic heart failure. J. Pathol. 1999, 188, 213–219. [Google Scholar] [CrossRef]
- Bussmann, N.; Breatnach, C.; Levy, P.T.; McCallion, N.; Franklin, O.; El-Khuffash, A. Early Diastolic Dysfunction and Respiratory Morbidity in Premature Infants: An Observational Study. J. Perinatol. 2018, 38, 1205–1211. [Google Scholar] [CrossRef]
- Mohamed, A.; Leeson, P.; Lewandowski, A.J. Like sheep, like humans? Right ventricular remodelling in a preterm-born ovine model. J. Physiol. 2018, 596, 5505–5506. [Google Scholar] [CrossRef] [Green Version]
- Lewandowski, A.J.; Bradlow, W.M.; Augustine, D.; Davis, E.F.; Francis, J.; Singhal, A.; Lucas, A.; Neubauer, S.; McCormick, K.; Leeson, P. Right ventricular systolic dysfunction in young adults born preterm. Circulation 2013, 128, 713–720. [Google Scholar] [CrossRef] [Green Version]
- Steurer, M.A.; Nawaytou, H.; Guslits, E.; Colglazier, E.; Teitel, D.; Fineman, J.R.; Keller, R.L. Mortality in infants with bronchopulmonary dysplasia: Data from cardiac catheterization. Pediatr. Pulmonol. 2019, 54, 804–813. [Google Scholar] [CrossRef] [PubMed]
- Altit, G.; Bhombal, S.; Hopper, R.K.; Tacy, T.A.; Feinstein, J. Death or resolution: The “natural history” of pulmonary hypertension in bronchopulmonary dysplasia. J. Perinatol. 2019, 39, 415–425. [Google Scholar] [CrossRef] [PubMed]
- del Cerro, M.J.; Sabaté Rotés, A.; Cartón, A.; Deiros, L.; Bret, M.; Cordeiro, M.; Verdú, C.; Barrios, M.I.; Albajara, L.; Gutierrez-Larraya, F. Pulmonary hypertension in bronchopulmonary dysplasia: Clinical findings, cardiovascular anomalies and outcomes. Pediatr. Pulmonol. 2014, 49, 49–59. [Google Scholar] [CrossRef] [PubMed]
- Zettler, E.; Rivera, B.K.; Stiver, C.; Boe, B.; Cua, C.; Ball, M.K.; Smith, C.V.; Slaughter, J.L.; Chen, B.; Callahan, R.; et al. Primary pulmonary vein stenosis among premature infants with single-vessel disease. J. Perinatol. 2021, 41, 1621–1626. [Google Scholar] [CrossRef] [PubMed]
- Drossner, D.M.; Kim, D.W.; Maher, K.O.; Mahle, W.T. Pulmonary Vein Stenosis: Prematurity and Associated Conditions. Pediatrics 2008, 122, e656–e661. [Google Scholar] [CrossRef]
- Zaidi, A.H.; Yamada, J.M.; Miller, D.T.; McEnaney, K.; Ireland, C.; Roberts, A.E.; Gauvreau, K.; Jenkins, K.J.; Chen, M.H. Clinical Syndromic Phenotypes and the Potential Role of Genetics in Pulmonary Vein Stenosis. Children 2021, 8, 128. [Google Scholar] [CrossRef]
- Seale, A.N.; Webber, S.A.; Uemura, H.; Partridge, J.; Roughton, M.; Ho, S.Y.; McCarthy, K.P.; Jones, S.; Shaughnessy, L.; Sunnegardh, J.; et al. Pulmonary vein stenosis: The UK, Ireland and Sweden collaborative study. Heart 2009, 95, 1944–1949. [Google Scholar] [CrossRef]
- Quinonez, L.G.; Gauvreau, K.; Borisuk, M.; Ireland, C.; Marshall, A.M.; Mayer, J.E.; Jenkins, K.J.; Fynn-Thompson, F.E.; Baird, C.W. Outcomes of surgery for young children with multivessel pulmonary vein stenosis. J. Thorac. Cardiovasc. Surg. 2015, 150, 911–917. [Google Scholar] [CrossRef] [Green Version]
- Weismann, C.G.; Asnes, J.D.; Bazzy-Asaad, A.; Tolomeo, C.; Ehrenkranz, R.A.; Bizzarro, M.J. Pulmonary hypertension in preterm infants: Results of a prospective screening program. J. Perinatol. 2017, 37, 572–577. [Google Scholar] [CrossRef]
- Check, J.; Gotteiner, N.; Liu, X.; Su, E.; Porta, N.; Steinhorn, R.; Mestan, K.K. Fetal growth restriction and pulmonary hypertension in premature infants with bronchopulmonary dysplasia. J. Perinatol. 2013, 33, 553–557. [Google Scholar] [CrossRef]
- Lagatta, J.M.; Hysinger, E.B.; Zaniletti, I.; Wymore, E.M.; Vyas-Read, S.; Yallapragada, S.; Nelin, L.D.; Truog, W.E.; Padula, M.A.; Porta, N.F.M.; et al. The Impact of Pulmonary Hypertension in Preterm Infants with Severe Bronchopulmonary Dysplasia through 1 Year. J. Pediatr. 2018, 203, 218–224. [Google Scholar] [CrossRef] [PubMed]
- Vanderlaan, R.D.; Caldarone, C.A. Pulmonary Vein Stenosis: Incremental Knowledge Gains to Improve Outcomes. Children 2021, 8, 481. [Google Scholar] [CrossRef] [PubMed]
- Masaki, N.; Adachi, O.; Katahira, S.; Saiki, Y.; Horii, A.; Kawamoto, S.; Saiki, Y. Progression of vascular remodeling in pulmonary vein obstruction. J. Thorac. Cardiovasc. Surg. 2020, 160, 777–790. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Huang, W.; Liu, H.; Zheng, Y.; Liao, L. Mechanical stretching of pulmonary vein stimulates matrix metalloproteinase-9 and transforming growth factor-β1 through stretch-activated channel/MAPK pathways in pulmonary hypertension due to left heart disease model rats. PLoS ONE 2020, 15, e0235824. [Google Scholar] [CrossRef] [PubMed]
- Hammer, P.E.; McEnaney, K.; Callahan, R.; Baird, C.W.; Hoganson, D.M.; Jenkins, K.J. The Role of Elevated Wall Shear Stress in Progression of Pulmonary Vein Stenosis: Evidence from Two Case Studies. Children 2021, 8, 729. [Google Scholar] [CrossRef]
- Niccum, M.; Callahan, R.; Gauvreau, K.; Jenkins, K.J. Aspiration Is Associated with Poor Treatment Response in Pediatric Pulmonary Vein Stenosis. Children 2021, 8, 783. [Google Scholar] [CrossRef]
- Xie, L.; Xiao, T.; Shen, J. Primary pulmonary vein stenosis in a premature infant without bronchopulmonary dysplasia: A case report. Heart Lung 2014, 43, 367–370. [Google Scholar] [CrossRef]
- Zozaya, C.; García González, I.; Avila-Alvarez, A.; Oikonomopoulou, N.; Sánchez Tamayo, T.; Salguero, E.; Saenz de Pipaón, M.; García-Muñoz Rodrigo, F.; Couce, M.L. Incidence, Treatment, and Outcome Trends of Necrotizing Enterocolitis in Preterm Infants: A Multicenter Cohort Study. Front. Pediatr. 2020, 8, 188. [Google Scholar] [CrossRef]
- Cotten, C.M. Modifiable Risk Factors in Necrotizing Enterocolitis. Clin. Perinatol. 2019, 46, 129–143. [Google Scholar] [CrossRef]
- Riedlinger, W.F.J.; Juraszek, A.L.; Jenkins, K.J.; Nugent, A.W.; Balasubramanian, S.; Calicchio, M.L.; Kieran, M.W.; Collins, T. Pulmonary vein stenosis: Expression of receptor tyrosine kinases by lesional cells. Cardiovasc. Pathol. 2006, 15, 91–99. [Google Scholar] [CrossRef]
- Hall, S.M.; Hislop, A.A.; Haworth, S.G. Origin, differentiation, and maturation of human pulmonary veins. Am. J. Respir. Cell Mol. Biol. 2002, 26, 333–340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sankar, M.N.; Bhombal, S.; Benitz, W.E. PDA: To treat or not to treat. Congenit. Heart Dis. 2019, 14, 46–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, C.; Gauvreau, K.; Levy, P.; Callahan, R.; Jenkins, K.J.; Chen, M. Longer Exposure to Left-to-Right Shunts Is a Risk Factor for Pulmonary Vein Stenosis in Patients with Trisomy 21. Children 2021, 8, 19. [Google Scholar] [CrossRef] [PubMed]
- Pierro, M.; Villamor-Martinez, E.; van Westering-Kroon, E.; Alvarez-Fuente, M.; Abman, S.H.; Villamor, E. Association of the dysfunctional placentation endotype of prematurity with bronchopulmonary dysplasia: A systematic review, meta-analysis and meta-regression. Thorax 2022, 77, 268–275. [Google Scholar] [CrossRef] [PubMed]
- Gowda, S.; Bhat, D.; Feng, Z.; Chang, C.H.; Ross, R.D. Pulmonary vein stenosis with Down syndrome: A rare and frequently fatal cause of pulmonary hypertension in infants and children. Congenit. Heart Dis. 2014, 9, E90–E97. [Google Scholar] [CrossRef]
- Martin, T.; Smith, A.; Breatnach, C.R.; Kent, E.; Shanahan, I.; Boyle, M.; Levy, P.T.; Franklin, O.; El-Khuffash, A. Infants Born with Down Syndrome: Burden of Disease in the Early Neonatal Period. J. Pediatr. 2018, 193, 21–26. [Google Scholar] [CrossRef]
- Shah, P.S.; Hellmann, J.; Adatia, I. Clinical characteristics and follow up of Down syndrome infants without congenital heart disease who presented with persistent pulmonary hypertension of newborn. J. Perinat. Med. 2004, 32, 168–170. [Google Scholar] [CrossRef]
- Smith, A.M.; Levy, P.T.; Franklin, O.; Molloy, E.; El-Khuffash, A. Pulmonary hypertension and myocardial function in infants and children with Down syndrome. Arch. Dis. Child. 2020, 105, 1031–1034. [Google Scholar] [CrossRef]
- van de Laar, I.; Wessels, M.; Frohn-Mulder, I.; Dalinghaus, M.; de Graaf, B.; van Tienhoven, M.; van der Moer, P.; Husen-Ebbinge, M.; Lequin, M.; Dooijes, D.; et al. First locus for primary pulmonary vein stenosis maps to chromosome 2q. Eur. Heart J. 2009, 30, 2485–2492. [Google Scholar] [CrossRef] [Green Version]
- Har-Even Cohn, R.; Hicks, M.; Lacson, A.; Hicks, A. Left hypoplastic lung and hemoptysis-rare familial unilateral pulmonary vein atresia. Clin. Case Rep. 2020, 8, 1698–1703. [Google Scholar] [CrossRef]
- Latson, L.A.; Prieto, L.R. Congenital and acquired pulmonary vein stenosis. Circulation 2007, 115, 103–108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holt, D.B.; Moller, J.H.; Larson, S.; Johnson, M.C. Primary pulmonary vein stenosis. Am. J. Cardiol. 2007, 99, 568–572. [Google Scholar] [CrossRef] [PubMed]
- Cory, M.J.; Ooi, Y.K.; Kelleman, M.S.; Vincent, R.N.; Kim, D.W.; Petit, C.J. Reintervention Is Associated With Improved Survival in Pediatric Patients With Pulmonary Vein Stenosis. JACC Cardiovasc. Interv. 2017, 10, 1788–1798. [Google Scholar] [CrossRef] [PubMed]
- Lo Rito, M.; Gazzaz, T.; Wilder, T.J.; Vanderlaan, R.D.; Van Arsdell, G.S.; Honjo, O.; Yoo, S.-J.; Caldarone, C.A. Pulmonary vein stenosis: Severity and location predict survival after surgical repair. J. Thorac. Cardiovasc. Surg. 2016, 151, 657–666.e2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalfa, D.; Belli, E.; Bacha, E.; Lambert, V.; di Carlo, D.; Kostolny, M.; Salminen, J.; Nosal, M.; Poncelet, A.; Horer, J.; et al. Primary Pulmonary Vein Stenosis: Outcomes, Risk Factors, and Severity Score in a Multicentric Study. Ann. Thorac. Surg. 2017, 104, 182–189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Callahan, R.; Esch, J.J.; Wang, G.; Ireland, C.M.; Gauvreau, K.; Jenkins, K.J. Systemic Sirolimus to Prevent In-Stent Stenosis in Pediatric Pulmonary Vein Stenosis. Pediatr. Cardiol. 2020, 41, 282–289. [Google Scholar] [CrossRef] [PubMed]
- Patel, J.D.; Briones, M.; Mandhani, M.; Jones, S.; Suthar, D.; Gray, R.; Pettus, J.; McCracken, C.; Thomas, A.; Petit, C.J. Systemic Sirolimus Therapy for Infants and Children With Pulmonary Vein Stenosis. J. Am. Coll. Cardiol. 2021, 77, 2807–2818. [Google Scholar] [CrossRef]
- Krishnan, U.; Feinstein, J.A.; Adatia, I.; Austin, E.D.; Mullen, M.P.; Hopper, R.K.; Hanna, B.; Romer, L.; Keller, R.L.; Fineman, J.; et al. Evaluation and Management of Pulmonary Hypertension in Children with Bronchopulmonary Dysplasia. J. Pediatr. 2017, 188, 24–34.e1. [Google Scholar] [CrossRef]
- Abman, S.H.; Hansmann, G.; Archer, S.L.; Ivy, D.D.; Adatia, I.; Chung, W.K.; Hanna, B.D.; Rosenzweig, E.B.; Raj, J.U.; Cornfield, D.; et al. Pediatric Pulmonary Hypertension: Guidelines From the American Heart Association and American Thoracic Society. Circulation 2015, 132, 2037–2099. [Google Scholar] [CrossRef]
- Kuo, J.A.; Petit, C.J. Pulmonary Vein Stenosis in Children: A Programmatic Approach Employing Primary and Anatomic Therapy. Children 2021, 8, 663. [Google Scholar] [CrossRef]
- Frank, B.S.; Schäfer, M.; Grenolds, A.; Ivy, D.D.; Abman, S.H.; Darst, J.R. Acute Vasoreactivity Testing during Cardiac Catheterization of Neonates with Bronchopulmonary Dysplasia-Associated Pulmonary Hypertension. J. Pediatr. 2019, 208, 127–133. [Google Scholar] [CrossRef] [PubMed]
- Minich, L.L.; Tani, L.Y.; Breinholt, J.P.; Tuohy, A.M.; Shaddy, R.E. Complete follow-up echocardiograms are needed to detect stenosis of normally connecting pulmonary veins. Echocardiography 2001, 18, 589–592. [Google Scholar] [CrossRef] [PubMed]
- Kato, H.; Fu, Y.Y.; Zhu, J.; Wang, L.; Aafaqi, S.; Rahkonen, O.; Slorach, C.; Traister, A.; Leung, C.H.; Chiasson, D.; et al. Pulmonary vein stenosis and the pathophysiology of “upstream” pulmonary veins. J. Thorac. Cardiovasc. Surg. 2014, 148, 245–253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sena, L.; Callahan, R.; Sleeper, L.A.; Beroukhim, R.S. Prognostic Significance of Computed Tomography Findings in Pulmonary Vein Stenosis. Children 2021, 8, 402. [Google Scholar] [CrossRef] [PubMed]
- Scott, M.V.; Fujii, A.M.; Behrman, R.H.; Dillon, J.E. Diagnostic ionizing radiation exposure in premature patients. J. Perinatol. 2014, 34, 392–395. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vyas-Read, S.; Varghese, N.P.; Suthar, D.; Backes, C.; Lakshminrusimha, S.; Petit, C.J.; Levy, P.T. Prematurity and Pulmonary Vein Stenosis: The Role of Parenchymal Lung Disease and Pulmonary Vascular Disease. Children 2022, 9, 713. https://doi.org/10.3390/children9050713
Vyas-Read S, Varghese NP, Suthar D, Backes C, Lakshminrusimha S, Petit CJ, Levy PT. Prematurity and Pulmonary Vein Stenosis: The Role of Parenchymal Lung Disease and Pulmonary Vascular Disease. Children. 2022; 9(5):713. https://doi.org/10.3390/children9050713
Chicago/Turabian StyleVyas-Read, Shilpa, Nidhy P. Varghese, Divya Suthar, Carl Backes, Satyan Lakshminrusimha, Christopher J. Petit, and Philip T. Levy. 2022. "Prematurity and Pulmonary Vein Stenosis: The Role of Parenchymal Lung Disease and Pulmonary Vascular Disease" Children 9, no. 5: 713. https://doi.org/10.3390/children9050713