

Different Dental Manifestations in Sisters with the Same ALPL Gene Mutation: A Report of Two Cases


Abstract
:1. Introduction
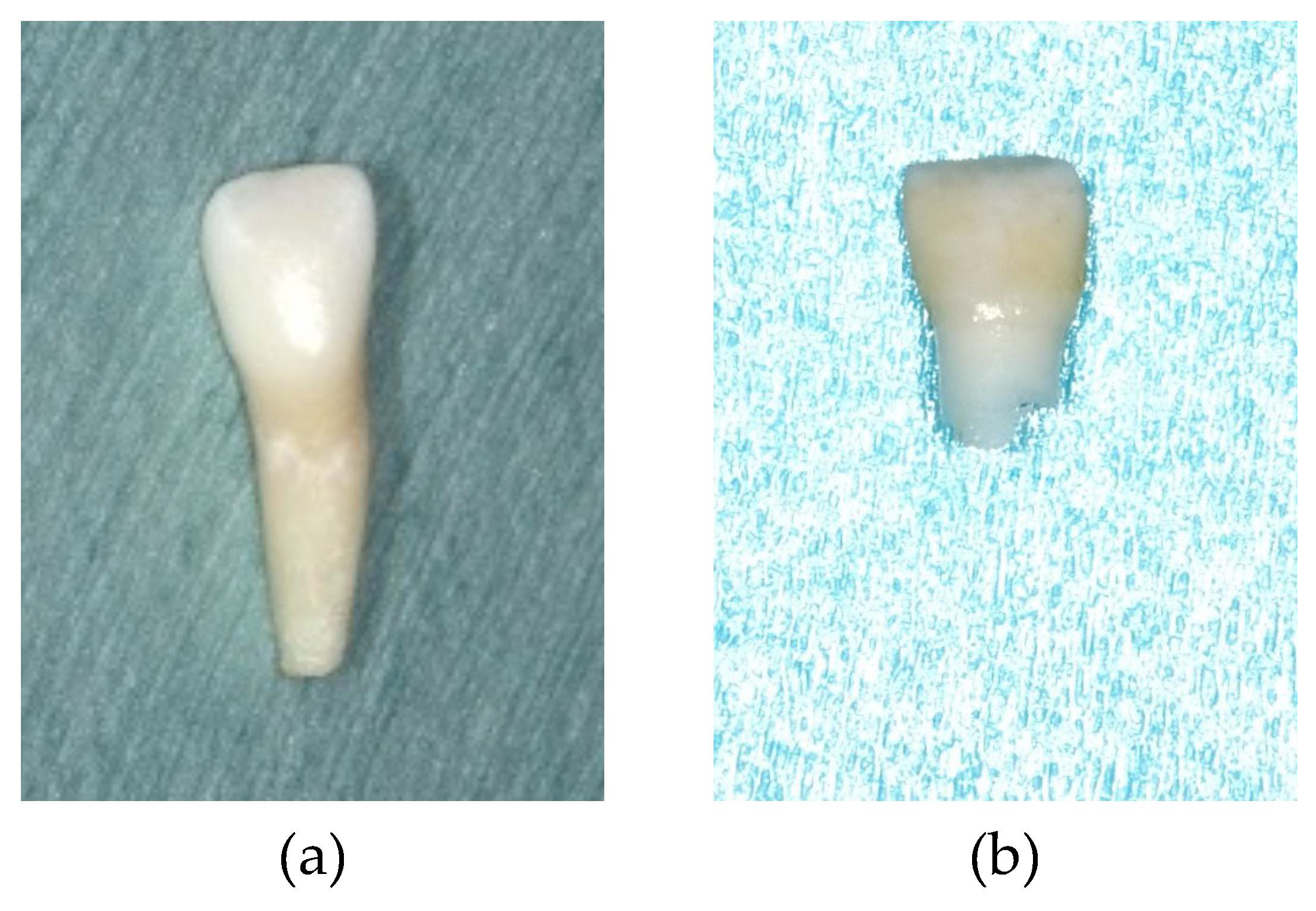
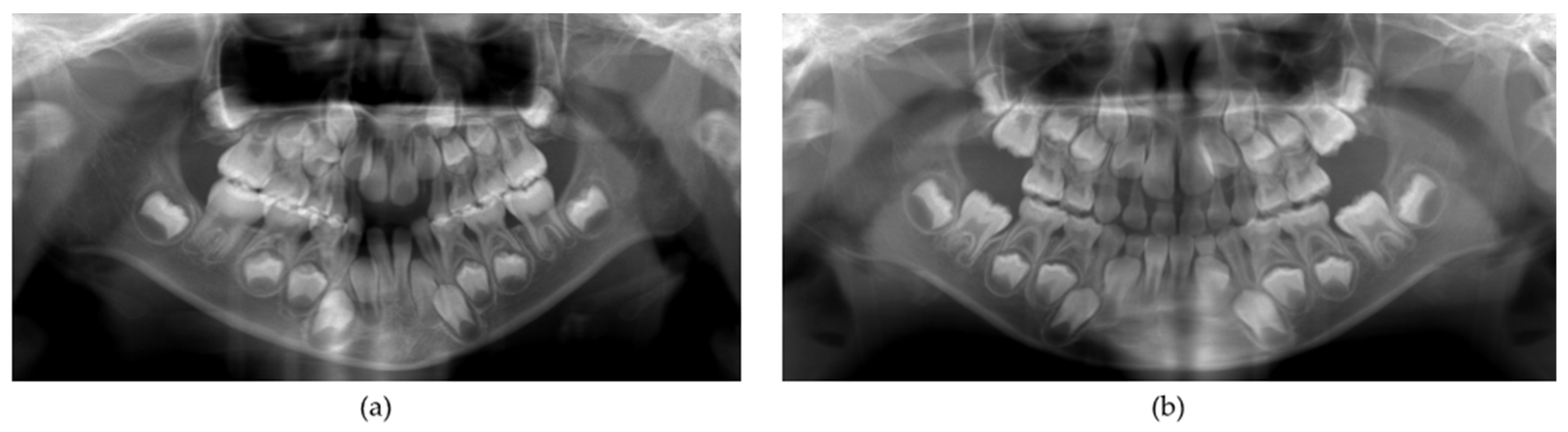
2. Case Report
3. Discussion
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Whyte, M.P. Physiological role of alkaline phosphatase explored in hypophosphatasia. Ann. N. Y. Acad. Sci. 2010, 1192, 190–200. [Google Scholar] [CrossRef]
- Rathbun, J.C. Hypophosphatasia; a new developmental anomaly. Am. J. Dis. Child. 1948, 75, 822–831. [Google Scholar] [CrossRef]
- Sultana, S.; Al-Shawafi, H.A.; Makita, S.; Sohda, M.; Amizuka, N.; Takagi, R.; Oda, K. An asparagine at position 417 of tissue-nonspecific alkaline phosphatase in essential for its structure and function as revealed by analysis of the N417S mutation associated with severe hypophosphatasia. Mol. Genet. Metab. 2013, 109, 282–288. [Google Scholar] [CrossRef]
- Kishnani, P.S.; Rush, E.T.; Arundel, P.; Bishop, N.; Dahir, K.; Fraser, W.; Harmatz, P.; Linglart, A.; Munns, C.F.; Nunes, M.E.; et al. Monitoring guidance for patients with hypophosphatasia treated with asfotase alfa. Mol. Genet. Metab. 2017, 122, 4–17. [Google Scholar] [CrossRef] [Green Version]
- Whyte, M.P. Atypical femoral fractures, bisphosphonates, and adult hypophosphatasia. J. Bone Miner. Res. 2009, 24, 1132–1134. [Google Scholar] [CrossRef]
- Okawa, R.; Kitaoka, T.; Saga, K.; Ozono, K.; Nakano, K. Report of two dental patients diagnosed with hypophosphatasia. J. Clin. Case Rep. 2016, 6, 1–5. [Google Scholar] [CrossRef] [Green Version]
- Okawa, R.; Nakano, K.; Matsumoto, M.; Kawabata, K.; Ooshima, T. Oral manifestations of patients with hypophosphatasia. Pediatric Dent. J. 2012, 22, 155–162. [Google Scholar] [CrossRef]
- Okawa, R.; Kadota, T.; Matayoshi, S.; Nakano, K. Dental manifestations leading to the diagnosis of hypophosphatasia in two children. J. Dent. Child. 2020, 87, 3–7. [Google Scholar]
- Bianchi, M.L.; Bishop, N.J.; Guañabens, N.; Hofmann, C.; Jakob, F.; Roux, C.; Zillikens, M.C.; Rare Bone Disease Action Group of the European Calcified Tissue Society. Hypophosphatasia in adolescents and adults: Overview of diagnosis and treatment. Osteoporos. Int. 2020, 31, 1445–1460. [Google Scholar] [CrossRef]
- Zierk, J.; Arzideh, A.; Haeckel, R.; Cario, H.; Frühwald, M.C.; Groß, H.; Gscheidmeier, T.; Hoffmann, R.; Krebs, A.; Lichtinghagen, R.; et al. Pediatric reference intervals for alkaline phosphatase. Clin. Chem. Lab. Med. 2017, 55, 102–110. [Google Scholar] [CrossRef]
- Saraff, V.; Narayanan, V.K.; Lawson, A.J.; Shaw, N.J.; Preece, M.A.; Högler, W. A diagnostic algorithm for children with low alkaline phosphatase activities: Lessons learned from laboratory screening for hypophosphatasia. J. Pediatr. 2016, 172, 181–186.e1. [Google Scholar] [CrossRef] [PubMed]
- Whyte, M.P. Hypophosphatasia—Aetiology nosology, pathogenesis, diagnosis and treatment. Nat. Rev. Endocrinol. 2016, 12, 233–246. [Google Scholar] [CrossRef] [PubMed]
- Mornet, E. Hypophosphatasia. Best Pract. Res. Clin. Rheumatol. 2008, 22, 113–127. [Google Scholar] [CrossRef] [PubMed]
- Whyte, M.P. Hypophosphatasia: An overview for 2017. Bone 2017, 102, 15–25. [Google Scholar] [CrossRef]
- Gomes-Filho, I.S.; Miranda, D.A.O.; Macêdo, T.C.N.; de Santana Passos, J. Relationship among gender, race, age, gingival width, and probing depth in primary teeth. J. Periodontol. 2006, 77, 1032–1042. [Google Scholar] [CrossRef]
- Kadota, T.; Okawa, R.; Otsugu, M.; Ohata, J.; Hanaoka, I.; Nakano, K. Mouthguards for a childhood hypophosphatasia patient to protect periodontal tissue of immature permanent teeth—Case report. Pediatr. Dent. J. 2021, 31, 117–122. [Google Scholar] [CrossRef]
- Weiss, M.J.; Ray, K.; Henthorn, P.S.; Lamb, B.; Kadesch, T.; Harris, H. Structure of the human liver/bone/kidney alkaline phosphatase gene. J. Biol. Chem. 1988, 263, 12002–12010. [Google Scholar] [CrossRef]
- Whyte, M.P.; Landt, M.; Ryan, L.M.; Henthorn, P.S.; Fedde, K.N.; Mahuren, J.D.; Coburn, S.P. Alkaline phosphatase: Placental and tissue-nonspecific isoenzymes hydrolyze phosphoethanolamine, inorganic pyrophosphate, and pyridoxal 5′-phosphate. Substrate accumulation in carriers of hypophosphatasia corrects during pregnancy. J. Clin. Investig. 1995, 95, 1440–1445. [Google Scholar] [CrossRef] [Green Version]
- Kato, M.; Michigami, T.; Tachikawa, K.; Kato, M.; Yabe, I.; Shimizu, T.; Asaka, T.; Kitagawa, Y.; Atsumi, T. Novel mutation in the ALPL gene with a dominant negative effect in a Japanese family. J. Bone Miner. Metab. 2021, 39, 804–809. [Google Scholar] [CrossRef]
- Lia-Baldini, A.S.; Muller, F.; Taillandier, A.; Gibrat, J.F.; Mouchard, M.; Robin, B.; Simon-Bouy, B.; Serre, J.L.; Aylsworth, A.S.; Bieth, E.; et al. A molecular approach to dominance in hypophosphatasia. Hum. Genet. 2001, 109, 99–108. [Google Scholar] [CrossRef]
- Whyte, M.P. Hypophosphatasia: Enzyme replacement therapy brings new opportunities and new challenges. J. Bone Miner. Res. 2017, 32, 667–675. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, T.; Mussawy, H.; Rolvien, T.; Hawellek, T.; Hubert, J.; Rüther, W.; Amling, M.; Barvencik, F. Clinical, radiographic and biochemical characteristics of adult hypophosphatasia. Osteoporos. Int. 2017, 28, 2653–2662. [Google Scholar] [CrossRef] [PubMed]
- Mornet, E.; Beck, C.; Bloch-Zupan, A.; Girschick, H.; Merrer, M.L. Clinical utility gene card for: Hypophosphatasia. Eur. J. Hum. Genet. 2011, 19, PMC3061990. [Google Scholar]
- Fraser, D. Hypophosphatasia. Am. J. Med. 1957, 22, 730–746. [Google Scholar] [CrossRef] [PubMed]
- Mornet, E. Hypophosphatasia. Orphanet J. Rare Dis. 2007, 2, 40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Atar, M.; Körperich, E.J. Systematic disorders and their influence on the development of dental hard tissues: A literature review. J. Dent. 2010, 38, 296–306. [Google Scholar] [CrossRef] [PubMed]
- Nakamura-Utsunomiya, A.; Okada, S.; Hara, K.; Miyagawa, S.; Takeda, K.; Fukuhara, R.; Nakata, Y.; Hayashidani, M.; Tachikawa, K.; Michigami, T.; et al. Clinical characteristics of perinatal lethal hypophosphatasia: A report of 6 cases. Clin. Pediatr. Endocrinol. 2010, 19, 7–13. [Google Scholar]
- Whyte, M.P.; Greenberg, C.R.; Salman, N.J.; Bober, M.B.; McAlister, W.H.; Wenkert, D.; Sickle, B.J.V.; Simmons, J.H.; Edgar, T.S.; Bauer, M.L.; et al. Enzyme-replacement therapy in life-threatening hypophosphatasia. N. Engl. J. Med. 2012, 366, 904–913. [Google Scholar]
- Okazaki, Y.; Kitajima, H.; Mochizuki, N.; Kitaoka, T.; Michigami, T.; Ozono, K. Lethal hypophosphatasia successfully treated with enzyme replacement from day 1 after birth. Eur. J. Pediatr. 2016, 175, 433–437. [Google Scholar]
- Müller, H.L.; Yamazaki, M.; Michigami, T.; Kageyama, T.; Schönau, E.; Schneider, P.; Ozono, K. Asp361Val Mutant of phosphatase found in patients with dominantly inherited hypophosphatasia inhibits the activity of the wild-type enzyme. J. Clin. Endocrinol. Metab. 2000, 85, 743–747. [Google Scholar] [CrossRef] [Green Version]
- Mornet, E. Genetics of hypophosphatasia. Arch. Pediatr. 2017, 24, 5S51–5S56. [Google Scholar] [CrossRef] [PubMed]
- Mornet, E. Molecular genetics of hypophosphatasia and phenotype-genotype correlations. In Neuronal Tissue-Nonspecific Alkaline Phosphatase (TNAP); Subcellular Biochemistry; Springer: Dordrecht, The Netherlands, 2015; Volume 76, pp. 25–43. [Google Scholar]
- Mornet, E.; Taillandier, A.; Domingues, C.; Dufour, A.; Benaloun, E.; Lavaud, N.; Wallon, F.; Rousseau, N.; Charle, C.; Guberto, M.; et al. Hypophosphatasia: A genetic-based nosology and new insights in genotype-phenotype correlation. Eur. J. Hum. Genet. 2021, 29, 289–299. [Google Scholar] [CrossRef] [PubMed]
- Beck, C.; Morbach, H.; Stenzel, M.; Collmann, H.; Schneider, P.; Girschick, H.J. Hypophosphatasia—Recent advances in di-agnosis and treatment. Open Bone J. 2009, 1, 8–15. [Google Scholar]
- Turan, S.; Topcu, B.; Gökçe, I.; Güran, T.; Atay, Z.; Omar, A.; Akçay, T.; Bereket, A. Serum alkaline phosphatase levels in healthy children and evaluation of alkaline phosphatase z-scores in different types of rickets. J. Clin. Res. Pediatr. Endocrinol. 2011, 3, 7–11. [Google Scholar] [CrossRef]
- Girschick, H.J.; Schneider, P.; Kruse, K.; Huppertz, H.I. Bone metabolism and bone mineral density in childhood hypophosphatasia. Bone 1999, 25, 361–367. [Google Scholar] [CrossRef]
- Okawa, R.; Iijima, O.; Kishino, M.; Okawa, H.; Toyosawa, S.; Tajima-Sugano, H.; Shimada, T.; Okada, T.; Ozono, K.; Ooshima, T.; et al. Gene therapy improved dental manifestations in hypophosphatasia model mice. J. Periodontal Res. 2017, 52, 471–478. [Google Scholar] [CrossRef]
- El-Labban, N.G.; Lee, K.W.; Rule, D. Permanent teeth in hypophosphatasia: Light and electron microscopic study. J. Oral Pathol. Med. 1991, 20, 352–360. [Google Scholar] [CrossRef]
- Olsson, A.; Matsson, L.; Blomquist, H.K.; Larsson, A.; Sjödin, B. Hypophosphatasia affecting the permanent dentition. J. Oral Pathol. Med. 1996, 25, 343–347. [Google Scholar] [CrossRef]
- Caton, J.G.; Armitage, G.; Berglundh, T.; Chapple, I.L.C.; Jepsen, S.; Kornman, K.S.; Mealey, B.L.; Papapanou, P.N.; Sanz, M.; Tonetti, M.S. A new classification scheme for periodontal and peri-implant diseases and conditions—Introduction and key changes from the 1999 classification. J. Clin. Periodontol. 2018, 45 (Suppl. S20), S1–S8. [Google Scholar] [CrossRef]
- Weerheijm, K.L.; Duggal, M.; Mejàre, I.; Papagiannoulis, L.; Koch, G.; Martens, L.C.; Hallonsten, A.-L. Judgement criteria for molar incisor hypomineralisation (MIH) in epidemiologic studies: A summary of the European meeting on MIH held in Athens, 2003. Eur. J. Paediatr. Dent. 2003, 4, 110–113. [Google Scholar]
- Elhennawy, K.; Manton, D.J.; Crombie, F.; Zaslansky, P.; Radlanski, R.; Jost-Brinkmann, P.; Schwendicke, F. Structual, mechanical and chemical evaluation of molar-incisor hypomineralization-affected enamel: A systematic review. Arch. Oral Biol. 2017, 83, 272–281. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Margarit, M.; Catalá-Pizarro, M.; Montiel-Company, J.M.; Almerich-Silla, J.M. Epidemiologic study of molar-incisor hypomineralization in 8-year-old Spanish children. Int. J. Paediatr. Dent. 2014, 24, 14–22. [Google Scholar] [CrossRef]
- Souza, J.F.; Costa-Silva, C.M.; Jeremias, F.; Santos-Pinto, L.; Zuanon, A.C.C.; Cordeiro, R.C.L. Molar incisor hypomineralisation: Possible aetiological factors in children from urban and rural areas. Eur. Arch. Paediatr. Dent. 2012, 13, 164–170. [Google Scholar] [CrossRef] [PubMed]
- Laisi, S.; Ess, A.; Sahlberg, C.; Arvio, P.; Lukinmaa, P.; Alaluusua, S. Amoxicillin may cause molar incisor hypomineralization. J. Dent. Res. 2009, 88, 132–136. [Google Scholar] [CrossRef] [PubMed]
- Kumazawa, K.; Sawada, T.; Yanagisawa, T.; Shintani, S. Effect of single-dose amoxicillin on rat incisor odontogenesis: A morphological study. Clin. Oral Investig. 2012, 16, 835–842. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Symptoms | Older Sister | Younger Sister | |
|---|---|---|---|
| Medical symptoms | Short stature | + | − |
| Low serum ALP | + | − | |
| High urine phosphoethanolamine (PEA) | + | − | |
| Metaphyseal irregularities | + | − | |
| Gait disturbance | + | − | |
| Dental symptoms | Early exfoliation of primary teeth | + | − |
| Probing pocket depth over 4 mm | + | − | |
| Mobility of teeth | + | − | |
| Hypomineralization of enamel | − | + | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kadota, T.; Ochiai, M.; Okawa, R.; Nakano, K. Different Dental Manifestations in Sisters with the Same ALPL Gene Mutation: A Report of Two Cases. Children 2022, 9, 1850. https://doi.org/10.3390/children9121850
Kadota T, Ochiai M, Okawa R, Nakano K. Different Dental Manifestations in Sisters with the Same ALPL Gene Mutation: A Report of Two Cases. Children. 2022; 9(12):1850. https://doi.org/10.3390/children9121850
Chicago/Turabian StyleKadota, Tamami, Marin Ochiai, Rena Okawa, and Kazuhiko Nakano. 2022. "Different Dental Manifestations in Sisters with the Same ALPL Gene Mutation: A Report of Two Cases" Children 9, no. 12: 1850. https://doi.org/10.3390/children9121850