
Rare Genetic Syndromes and Oral Anomalies: A Review of the Literature and Case Series with a New Classification Proposal

 ,
, 
Abstract
:1. Introduction
- Homogeneity: Each disease is defined by its clinical homogeneity, regardless of the etiology or the number of identified responsible genes.
- Rarity: Each disease is defined based on the European legislation, which establishes a prevalence threshold not exceeding 5 affected persons per 10,000 people.
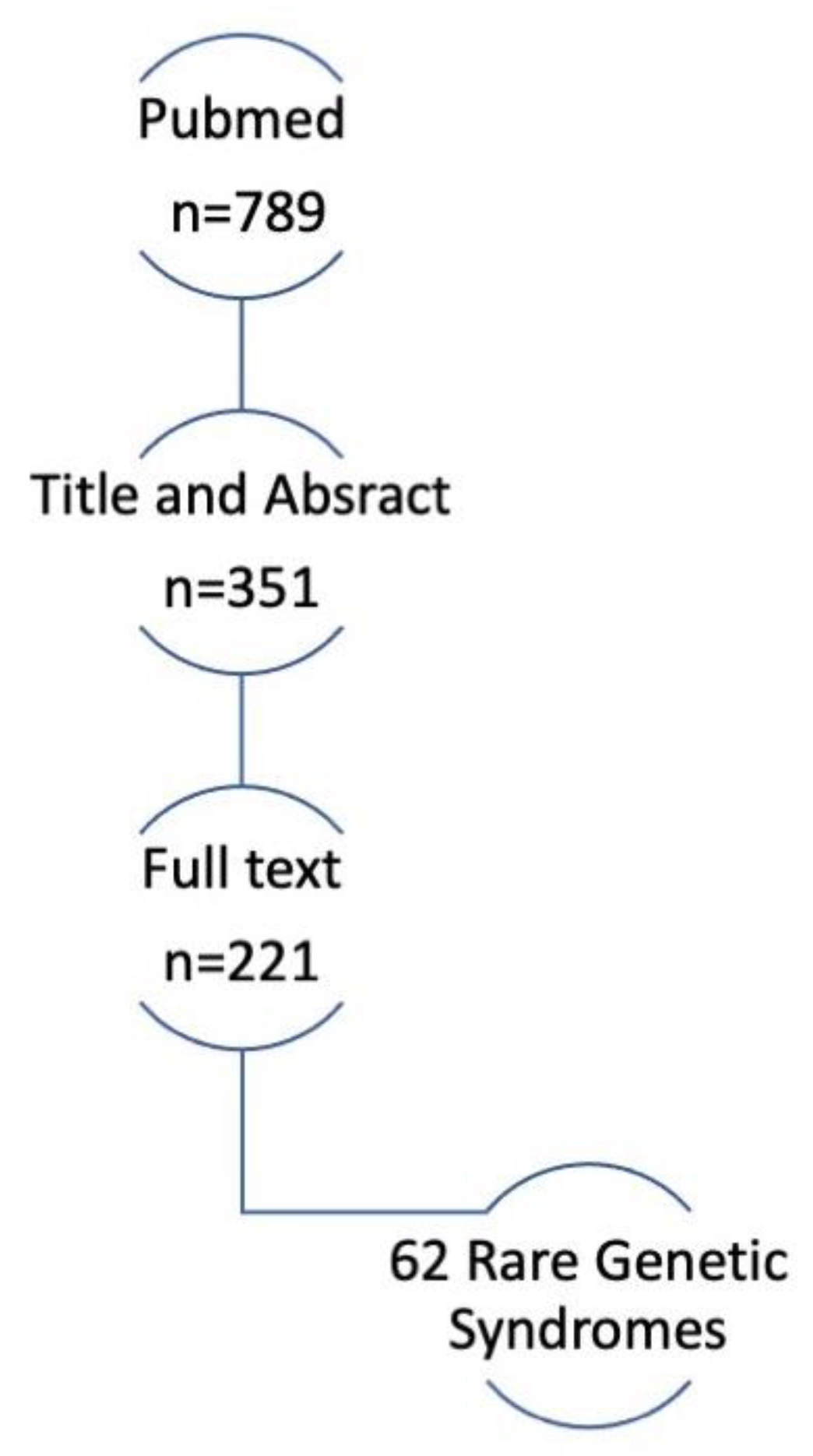
2. Materials and Methods
- Rare genetic syndromes including dental anomalies.
- Rare genetic syndromes including anomalies of the maxillary bones.
- Rare genetic syndromes including anomalies of the soft oral tissues.
- Rare genetic syndromes including mixed oral anomalies.
- They have focused on the genetic aspects only.
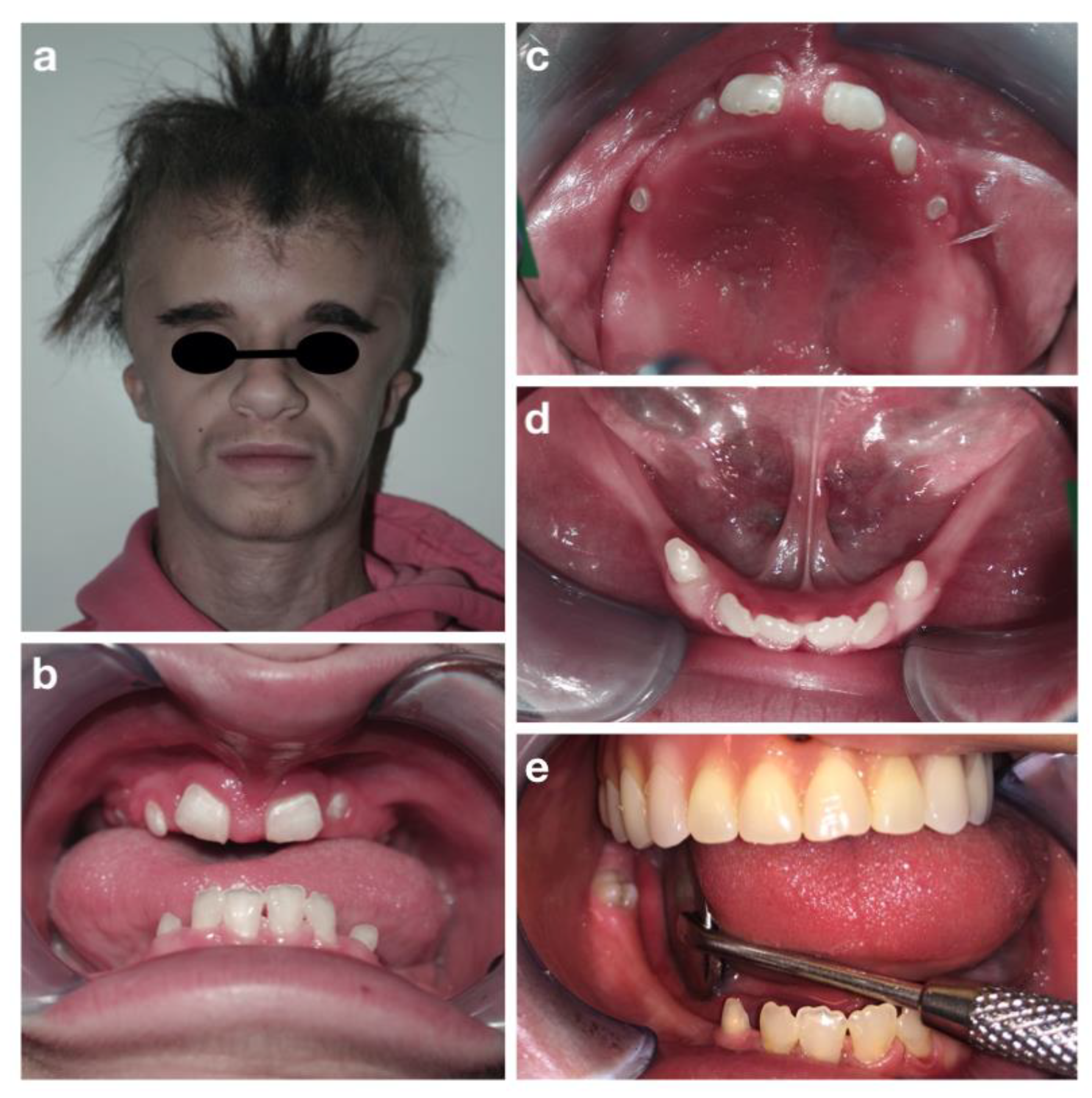
3. Results
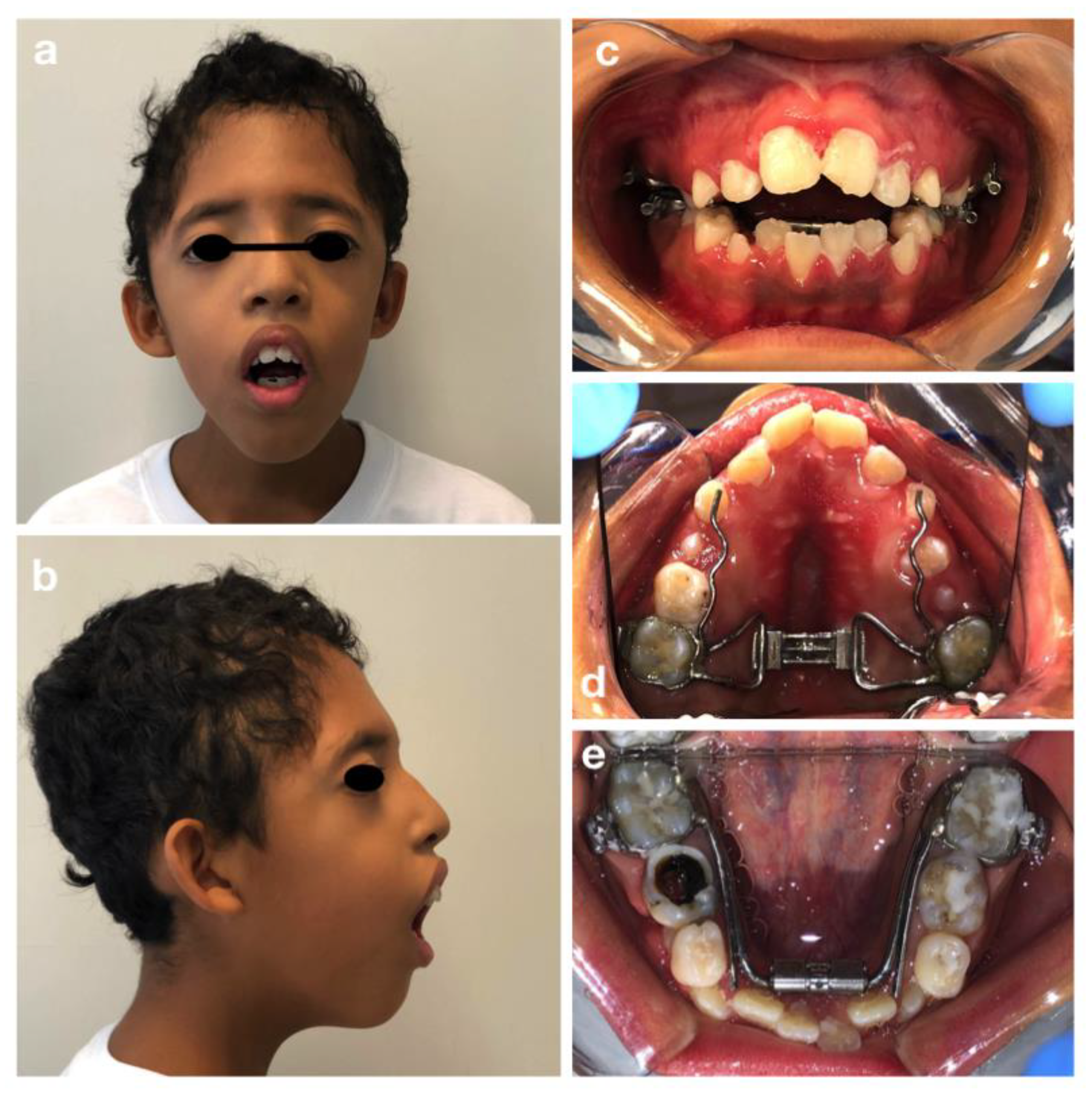
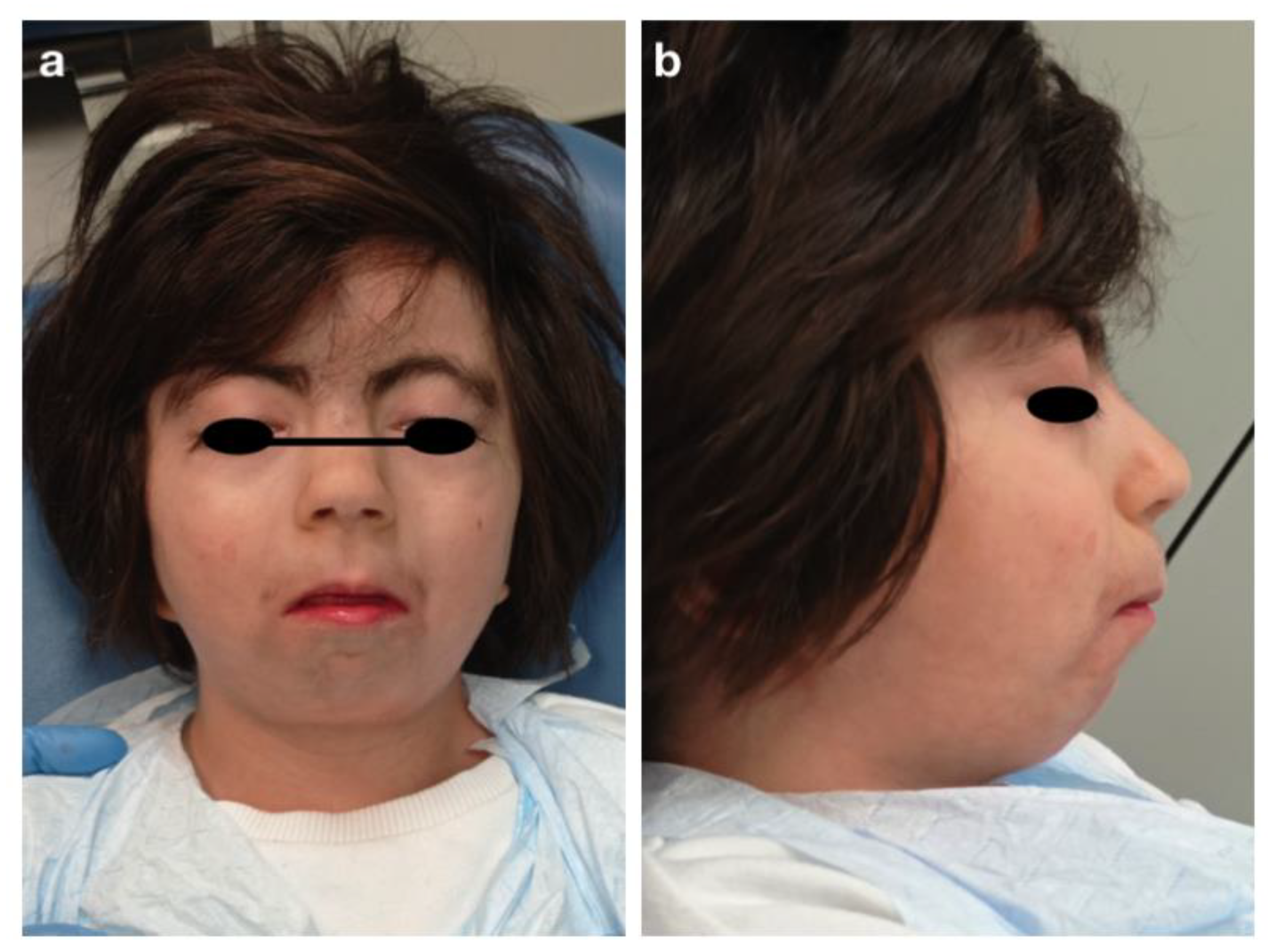
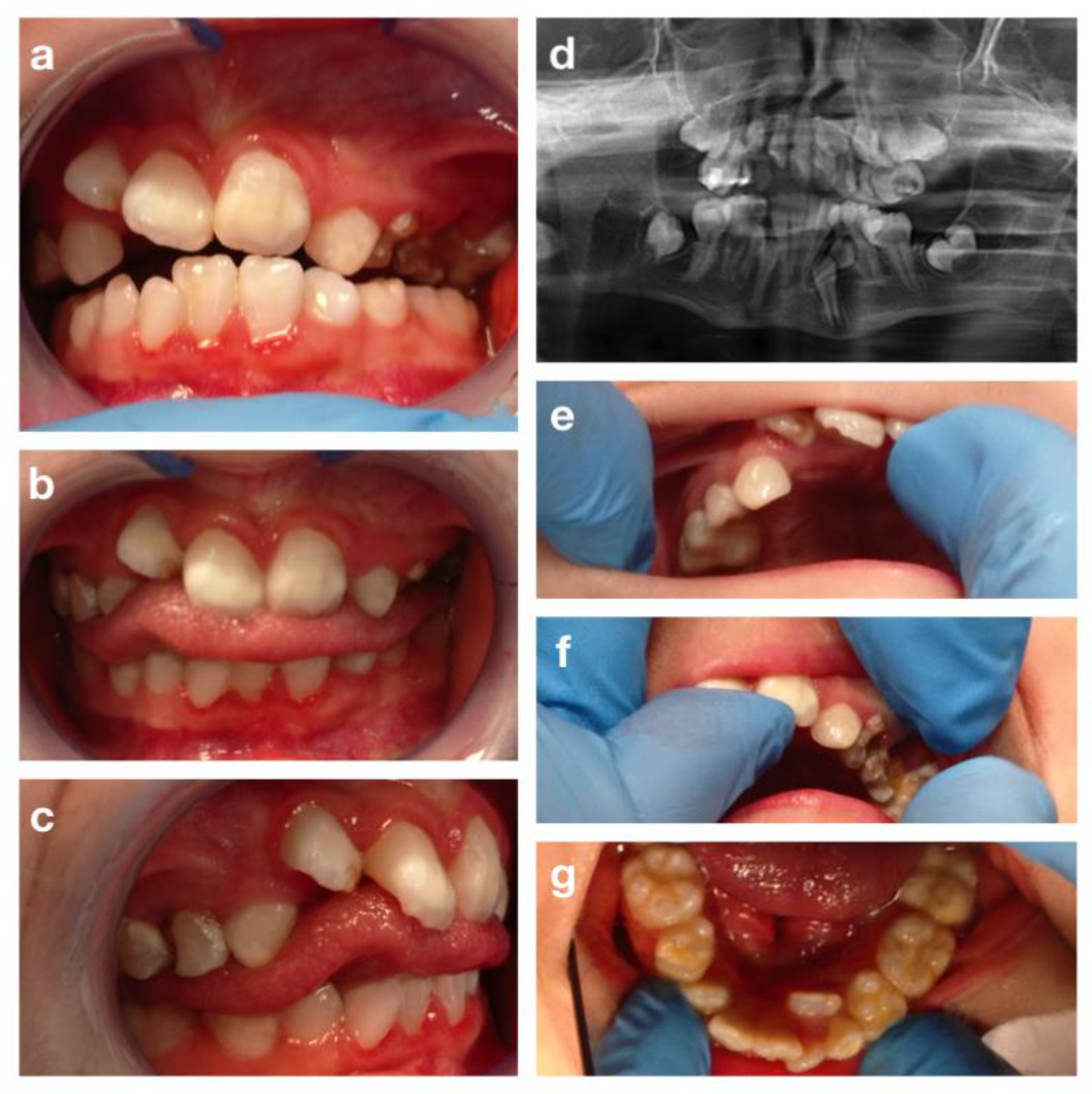
3.1. Crouzon Syndrome
3.2. Congenital Nemaline Myopathies
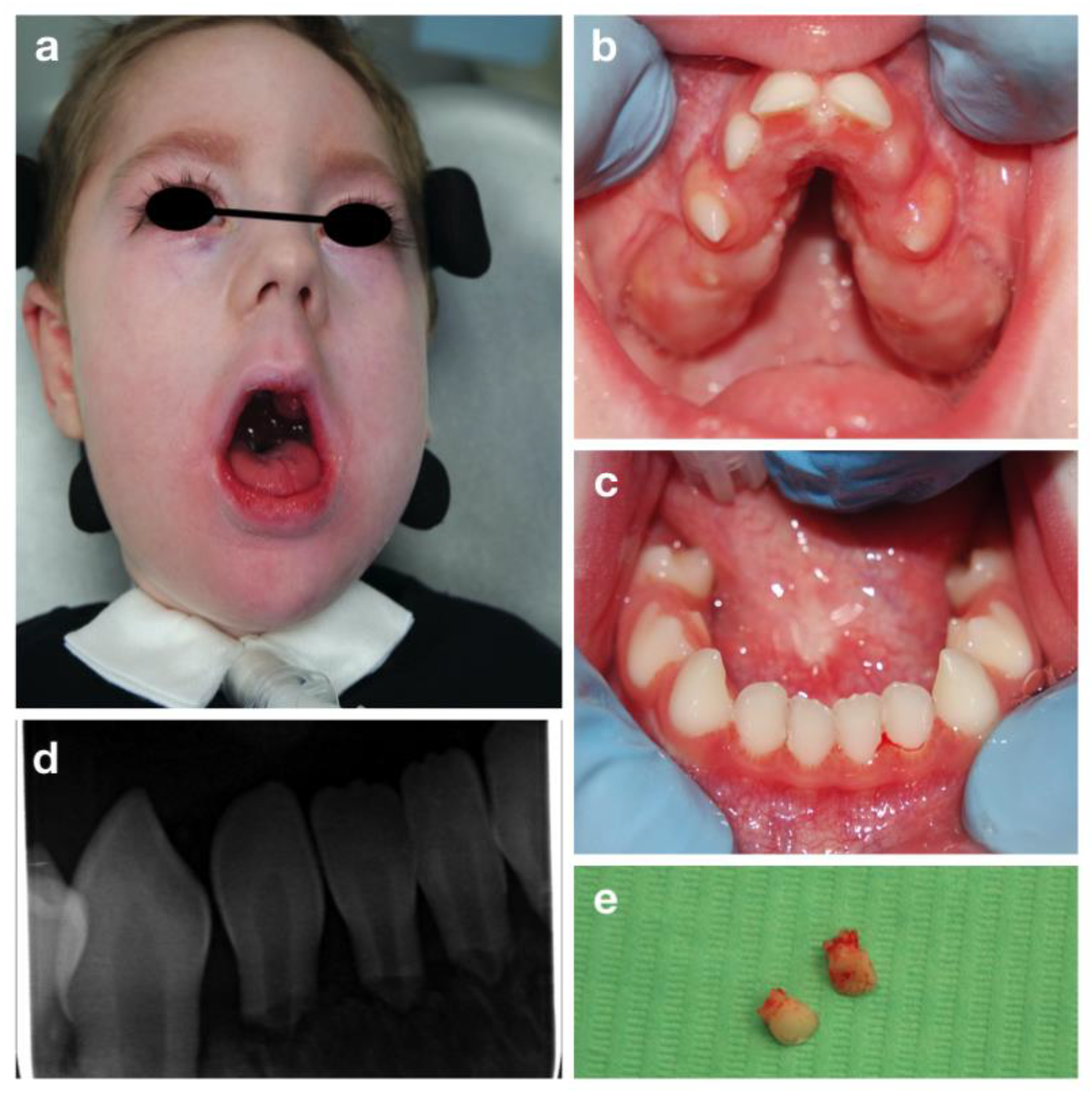
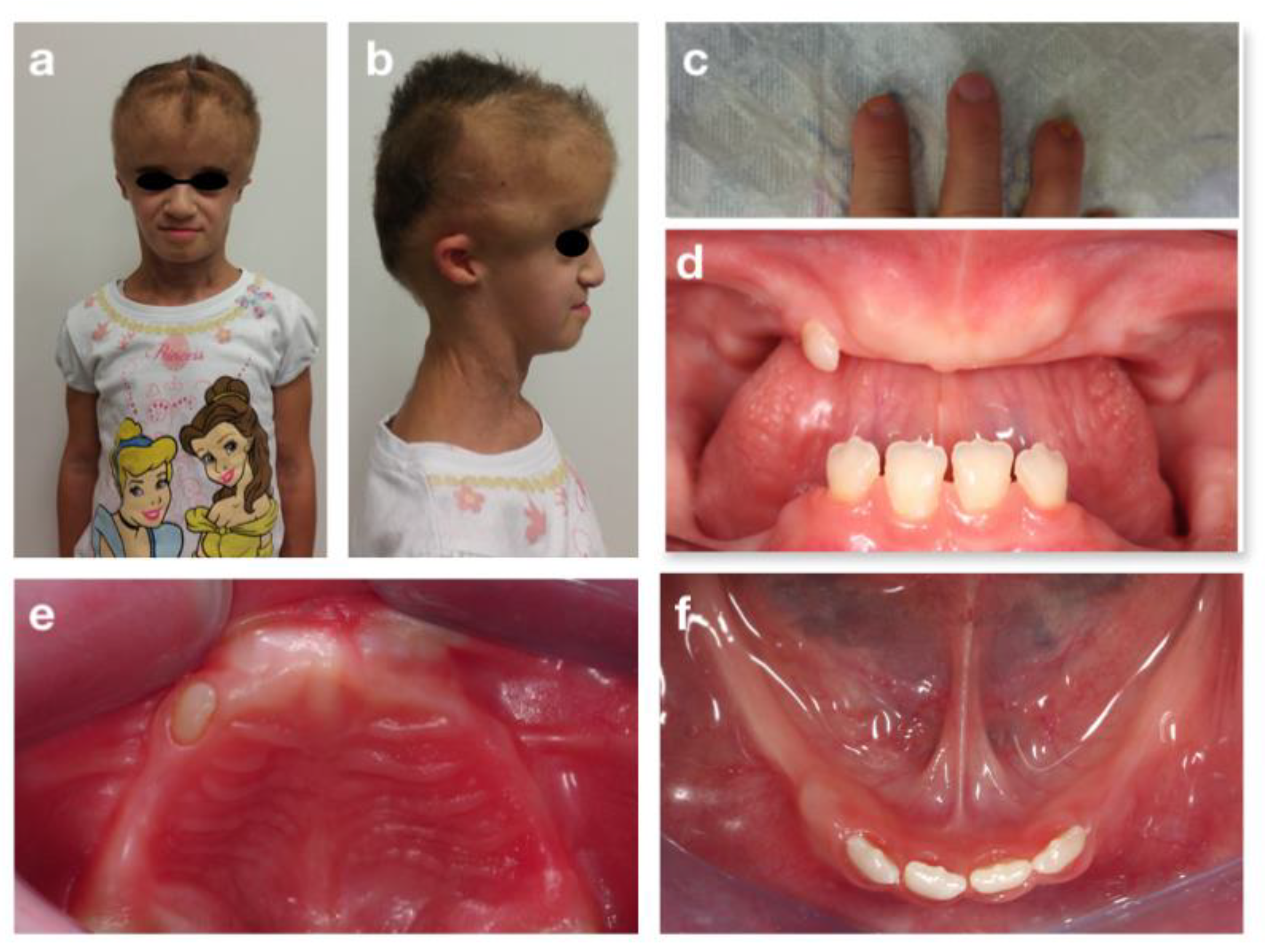
3.3. CHARGE Syndrome
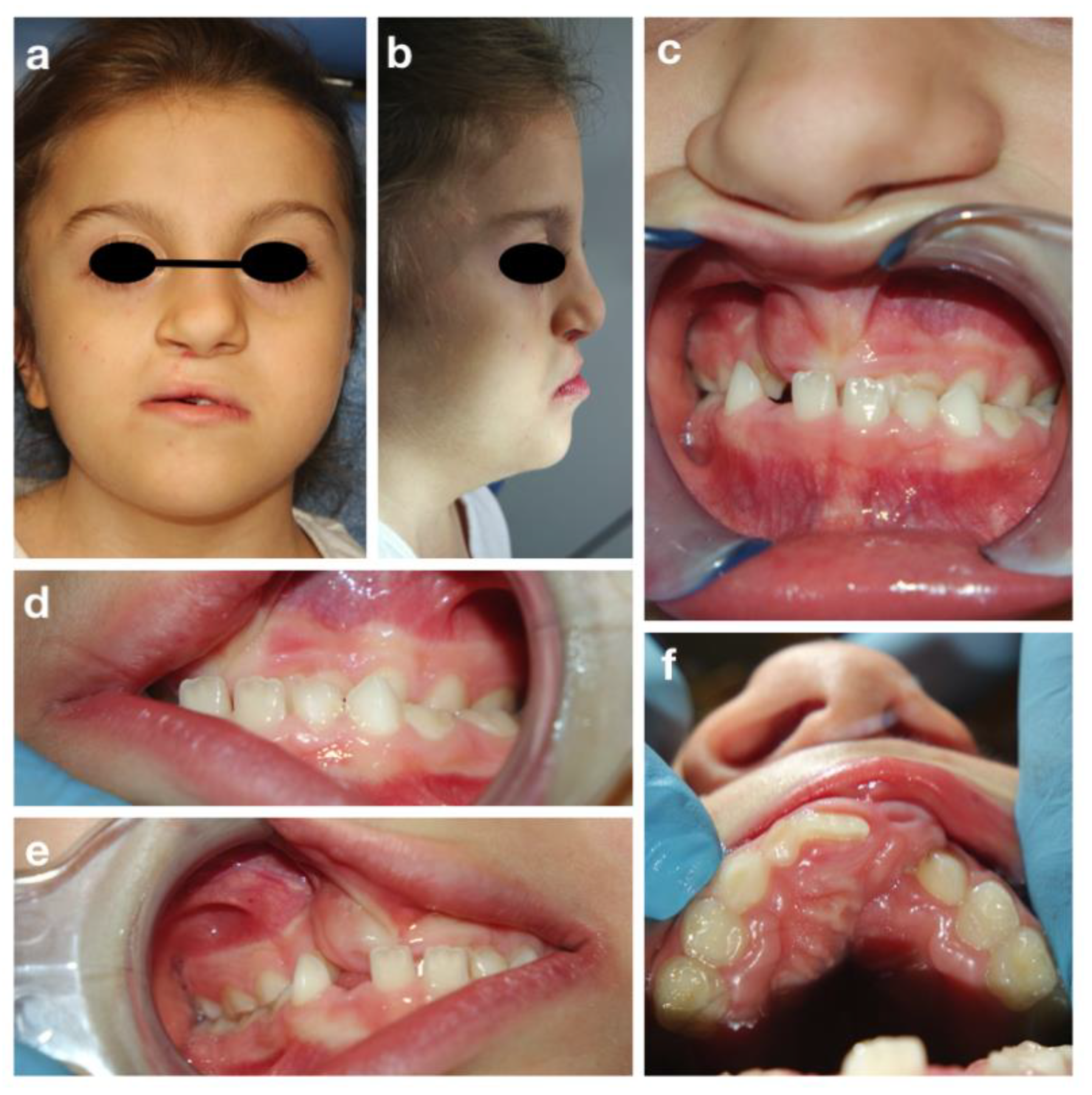
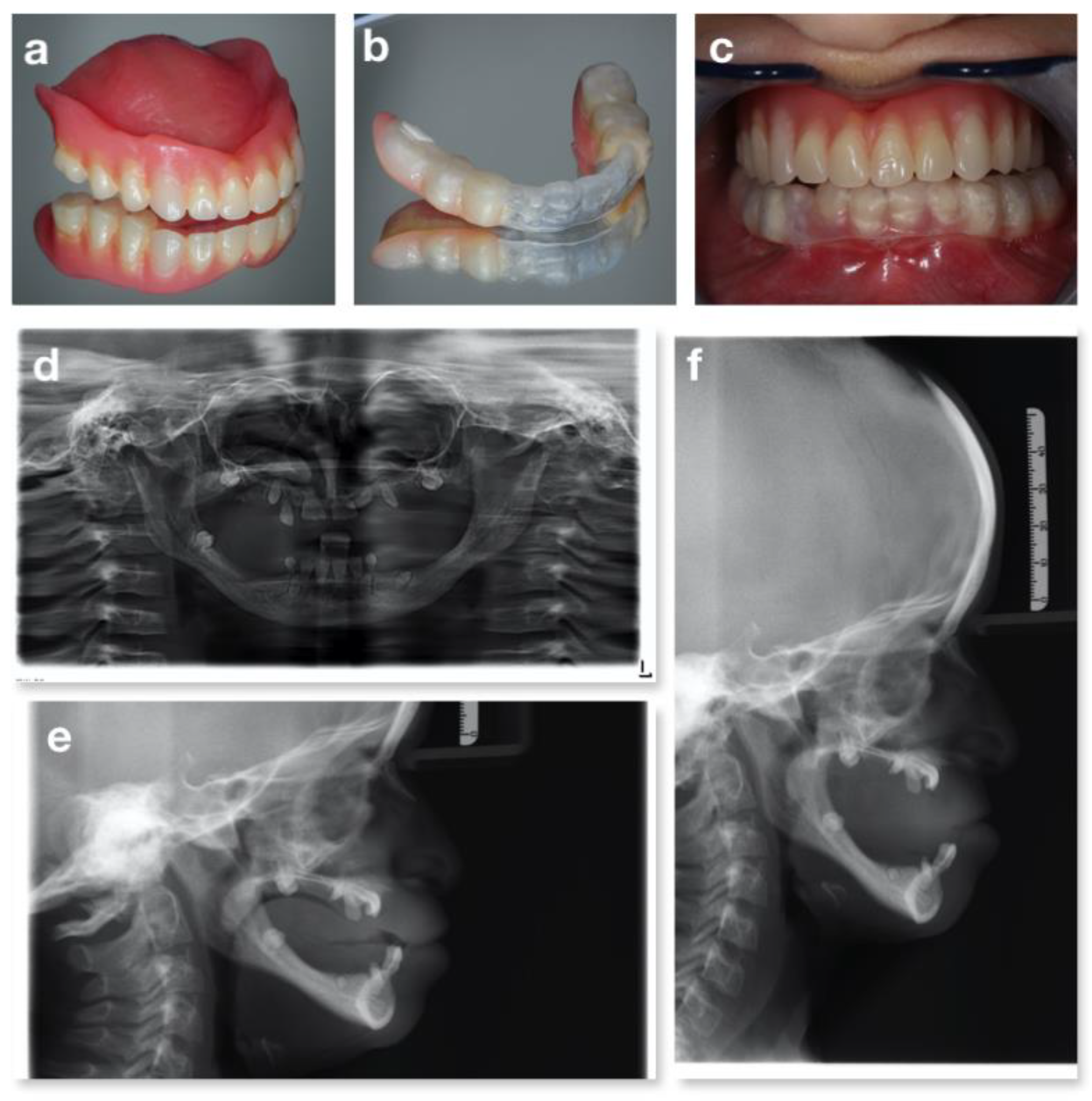
3.4. The Cornelia De Lange Syndrome
3.5. Progeroid Syndromes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Richter, T.; Nestler-Parr, S.; Babela, R.; Khan, Z.M.; Tesoro, T.; Molsen, E.; Hughes, D. Rare Disease Terminology and Definitions—A Systematic Global Review: Report of the ISPOR Rare Disease Special Interest Group. Value Health 2015, 18, 906–914. [Google Scholar] [CrossRef] [Green Version]
- European Project for Rare Diseases National Plans Management EUROPLAN. Available online: www.euro-planproject.eu/Content?folder=1 (accessed on 15 November 2021).
- Thesleff, I. The genetic basis of normal and abnormal craniofacial development. Acta Odontol. Scand. 1998, 56, 321–325. [Google Scholar] [CrossRef]
- London Dysmorphology Database. Available online: http://www.lmdatabases.com/ (accessed on 15 July 2021).
- London, J.; Birkedal-Hansen, H. Opportunities in dental, oral, and craniofacial research. Compend. Contin. Educ. Dent. 2000, 21, 760–762, 764, 766. [Google Scholar] [PubMed]
- Slavkin, H. Craniofacial-Oral-Dental Research in the 21st Century. J. Dent. Res. 1997, 76, 628–630. [Google Scholar] [CrossRef] [PubMed]
- Slavkin, H.C. Molecular Biology Experimental Strategies for Craniofacial-Oral-Dental Dysmorphology. Connect. Tissue Res. 1995, 32, 233–239. [Google Scholar] [CrossRef]
- Bloch-Zupan, A.; Sedano, H.; Scully, C. Dento/Oro/Craniofacial Anomalies and Genetics, 1st ed.; Elsevier: Amsterdam, The Netherlands, 2012; pp. 1–8. [Google Scholar]
- Alliot-Licht, B.; Lusson, C.; Hyon, I.; Dajean-Trutaud, S.; Le Caignec, C.; Lopez-Cazaux, S. Signes extra-oraux à rechercher face à des signes bucco-dentaires d’alerte de maladies d’origine génétique [Extra-oral signs to look for in patients exhibiting oral warning signs of genetic diseases]. Comptes Rendus Biol. 2015, 338, 48–57. [Google Scholar] [CrossRef] [PubMed]
- Boycott, K.M.; Rath, A.; Chong, J.; Hartley, T.; Alkuraya, F.S.; Baynam, G.; Brookes, A.J.; Brudno, M.; Carracedo, A.; Dunnen, J.T.D.; et al. International Cooperation to Enable the Diagnosis of All Rare Genetic Diseases. Am. J. Hum. Genet. 2017, 100, 695–705. [Google Scholar] [CrossRef] [Green Version]
- American Academy of Pediatric Dentistry. Guideline on dental management of heritable dental developmental anomalies. Pediatr. Dent. 2013, 35, E179–E184. [Google Scholar]
- Orpha.net. Available online: https://www.orpha.net/ (accessed on 17 July 2021).
- Phenodent. Available online: http://www.phenodent.org/ (accessed on 17 July 2021).
- GARD. Available online: https://rarediseases.info.nih.gov/ (accessed on 17 July 2021).
- Online Mendelian Inheritance in Man. Available online: https://omim.org/ (accessed on 11 November 2020).
- Amberger, J.S.; Bocchini, C.A.; Schiettecatte, F.; Scott, A.F.; Hamosh, A. OMIM.org: Online Mendelian Inheritance in Man (OMIM®), an online catalog of human genes and genetic disorders. Nucleic Acids Res. 2015, 43, D789–D798. [Google Scholar] [CrossRef] [Green Version]
- Regulation (EC) n° 141/2000 of the European Parliament and of the Council of 16 December 1999 on Orphan Drugs. Available online: https://eur-lex.europa.eu/legal-content/EN/TXT/?uri=celex%3A32000R0141 (accessed on 15 November 2020).
- Kaushik, A.; Bhatia, H.; Sharma, N. Crouzon’s Syndrome: A Rare Genetic Disorder. Int. J. Clin. Pediatr. Dent. 2016, 9, 384–387. [Google Scholar] [CrossRef] [PubMed]
- Carinci, F.; Pezzetti, F.; Locci, P.; Becchetti, E.; Carls, F.; Avantaggiato, A.; Becchetti, A.; Carinci, P.; Baroni, T.; Bodo, M. Apert and Crouzon Syndromes: Clinical Findings, Genes and Extracellular Matrix. J. Craniofac. Surg. 2005, 16, 361–368. [Google Scholar] [CrossRef] [Green Version]
- Helman, S.N.; Badhey, A.; Kadakia, S.; Myers, E. Revisiting Crouzon syndrome: Reviewing the background and management of a multifaceted disease. Oral Maxillofac. Surg. 2014, 18, 373–379. [Google Scholar] [CrossRef] [PubMed]
- Khandelwal, R.; Agrawal, P.; Majumdar, M.R. Crouzon syndrome. BMJ Case Rep. 2012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crouzon, O. Dysostose Cranio-Faciale Héréditaire; Bulletins et mémoires de la Société Médicale des Hôpitaux: Paris, France, 1912; p. 545. [Google Scholar]
- Renier, D.; Lajeunie, E.; Arnaud, E.; Marchac, D. Management of craniosynostoses. Child’s Nerv. Syst. 2000, 16, 645–658. [Google Scholar] [CrossRef]
- Khominsky, A.; Yong, R.; Ranjitkar, S.; Townsend, G.; Anderson, P.J. Extensive phenotyping of the orofacial and dental complex in Crouzon syndrome. Arch. Oral Biol. 2018, 86, 123–130. [Google Scholar] [CrossRef] [PubMed]
- Staal, F.C.; Ponniah, A.J.; Angullia, F.; Ruff, C.; Koudstaal, M.; Dunaway, D. Describing Crouzon and Pfeiffer syndrome based on principal component analysis. J. Cranio-Maxillofac. Surg. 2015, 43, 528–536. [Google Scholar] [CrossRef]
- Claeys, K.G. Congenital myopathies: An update. Dev. Med. Child Neurol. 2020, 62, 297–302. [Google Scholar] [CrossRef]
- Sewry, C.A.; Laitila, J.M.; Wallgren-Pettersson, C. Nemaline myopathies: A current view. J. Muscle Res. Cell Motil. 2019, 40, 111–126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anderson, P.J.; Barker, J.H.; David, D.J. Management of facial dysmorphogenesis in nemaline myopathy: A case re-port. World J. Orthod. 2005, 6, 156–160. [Google Scholar]
- Bergman, J.; Janssen, N.; Hoefsloot, L.H.; Jongmans, M.C.; Hofstra, R.; Van Ravenswaaij-Arts, C.M.A. CHD7 mutations and CHARGE syndrome: The clinical implications of an expanding phenotype. J. Med Genet. 2011, 48, 334–342. [Google Scholar] [CrossRef]
- Usman, N.; Sur, M. CHARGE Syndrome. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2021. [Google Scholar]
- Hudson, A.; Trider, C.-L.; Blake, K. CHARGE Syndrome. Pediatr. Rev. 2017, 38, 56–59. [Google Scholar] [CrossRef] [Green Version]
- Hsu, P.; Ma, A.; Wilson, M.; Williams, G.; Curotta, J.; Munns, C.F.; Mehr, S. CHARGE syndrome: A review. J. Paediatr. Child Health 2014, 50, 504–511. [Google Scholar] [CrossRef] [PubMed]
- Blake, K.D.; Davenport, S.L.H.; Hall, B.D.; Hefner, M.A.; Pagon, R.A.; Williams, M.S.; Lin, A.E.; Graham, J.M., Jr. CHARGE Association: An Update and Review for the Primary Pediatrician. Clin. Pediatr. 1998, 37, 159–173. [Google Scholar] [CrossRef] [PubMed]
- Sanlaville, D.; Verloes, A. CHARGE syndrome: An update. Eur. J. Hum. Genet. 2007, 15, 389–399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cascella, M.; Muzio, M.R. Cornelia de Lange Syndrome. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2021. [Google Scholar]
- Bergeron, M.; Bs, K.C.; Ishman, S.L. Cornelia de lange manifestations in otolaryngology: A systematic review and meta-analysis. Laryngoscope 2020, 130, E122–E133. [Google Scholar] [CrossRef]
- Boyle, M.; Jespersgaard, C.; Brøndum-Nielsen, K.; Bisgaard, A.-M.; Tümer, Z. Cornelia de Lange syndrome. Clin. Genet. 2015, 88, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Deardorff, M.A.; Noon, S.E.; Krantz, I.D. Cornelia de Lange Syndrome. In GeneReviews®; Adam, M.P., Noon, S.E., Krantz, I.D., Eds.; University of Washington: Seattle, DC, USA, 2005. [Google Scholar]
- Grau Carbó, J.; López Jiménez, J.; Giménez Prats, M.J.; Sànchez Molins, M. Cornelia de Lange syndrome: A case report. Med. Oral Patol. Oral Cir. Bucal 2007, 12, E445–E448. [Google Scholar]
- Guadagni, M.G.; Cetrullo, N.; Piana, G. Cornelia de Lange syndrome: Description of the orofacial features and case re-port. Eur. J. Paediatr. Dent. 2008, 9 (Suppl. 4), 9–13. [Google Scholar]
- Sandhu, M.; Nagpal, M.; Gulia, S.; Sachdev, V. Dental Management of Cornelia de Lange Syndrome: A Rare Case Report. J. Clin. Diagn. Res. 2015, 9, ZD12–ZD14. [Google Scholar] [CrossRef] [PubMed]
- Toker, A.S.; Ay, S.; Yeler, H.; Sezgin, I. Dental Findings in Cornelia De Lange Syndrome. Yonsei Med. J. 2009, 50, 289–292. [Google Scholar] [CrossRef] [Green Version]
- Sinha, J.K.; Ghosh, S.; Raghunath, M. Progeria: A rare genetic premature ageing disorder. Indian J. Med Res. 2014, 139, 667–674. [Google Scholar]
- Carrero, D.; Soria-Valles, C.; López-Otín, C. Hallmarks of progeroid syndromes: Lessons from mice and reprogrammed cells. Dis. Model. Mech. 2016, 9, 719–735. [Google Scholar] [CrossRef] [Green Version]
- Ullrich, N.J.; Silvera, V.M.; Campbell, S.E.; Gordon, L.B. Craniofacial Abnormalities in Hutchinson-Gilford Progeria Syndrome. Am. J. Neuroradiol. 2012, 33, 1512–1518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, S. Textbook of Periodontia, 3rd ed.; Blakiston Co.: Philadelphia, PA, USA, 1950. [Google Scholar]
- Tanwar, R.; Iyengar, A.R.; Nagesh, K.S.; Subhash, B. Crouzons syndrome: A case report with review of literature. J. Indian Soc. Pedod. Prev. Dent. 2013, 31, 118. [Google Scholar] [CrossRef]
- Cagetti, M.G.; Balian, A.; Cirio, S.; Camoni, N.; Salerno, C.; Tartaglia, G.M. Is Pediatric Dentistry a Topic of Interest for Pediatric Journals? A Scoping Review. Children 2021, 8, 720. [Google Scholar] [CrossRef]
- Ferrazzano, G.F.; Salerno, C.; Sangianantoni, G.; Caruso, S.; Ingenito, A.; Cantile, T. The Effect of Dental Treatment under General Anesthesia on Quality of Life and Growth and Blood Chemistry Parameters in Uncooperative Pediatric Patients with Compromised Oral Health: A Pilot Study. Int. J. Environ. Res. Public Health 2020, 17, 4407. [Google Scholar] [CrossRef] [PubMed]
- Nqcobo, C.; Ralephenya, T.; Kolisa, Y.M.; Esan, T.; Yengopal, V. Caregivers’ perceptions of the oral-health-related quality of life of children with special needs in Johannesburg, South Africa. Health SA 2019, 24, 1056. [Google Scholar] [CrossRef] [PubMed]
- D’Addazio, G.; Santilli, M.; Sinjari, B.; Xhajanka, E.; Rexhepi, I.; Mangifesta, R.; Caputi, S. Access to Dental Care—A Survey from Dentists, People with Disabilities and Caregivers. Int. J. Environ. Res. Public Health 2021, 18, 1556. [Google Scholar] [CrossRef] [PubMed]
- Pober, B.R. Williams–Beuren Syndrome. N. Engl. J. Med. 2010, 362, 239–252. [Google Scholar] [CrossRef] [PubMed]
- Ministero Della Salute, Raccomandazioni per la Promozione Della Salute Orale in età Perinatale. 2014. Available online: https://www.salute.gov.it/imgs/C_17_pubblicazioni_2317_allegato.pdf (accessed on 15 November 2021).
- Charteris, P.; Kinsella, T. The Oral Care Link Nurse: A facilitator and educator for maintaining oral health for patients at the Royal Hospital for neuro-disability. Spéc. Care Dent. 2001, 21, 68–71. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Type of Anomaly | Clinical Signs | Anomalies Observed |
|---|---|---|
| Number anomalies | ||
| Reduced number | Anodontia | Absence of all dental elements |
| Oligodontia | Presence of less than half of the normal number of teeth | |
| Ipodontia | Presence of more than half of the normal number of teeth | |
| Increased number | Supplementary teeth | The tooth repeats the shape and function of the adjacent tooth |
| Supernumerary teeth | Elements are atypical, smaller and rudimentary. They are classified into: | |
| -Mesiodens: between the two upper central incisors | ||
| -Paramolar: in the molar region | ||
| -Distomolar: behind the third molar | ||
| Location anomalies | Ectopia | The element is located near the usual site, in a vestibular, lingual or palatal position |
| Transposition | Two contiguous teeth reciprocally invert their position | |
| Heterotopia | Element located far from the usual position. If the position is outside the oral cavity it is defined as migration. | |
| Position anomalies | Version | Inclination of a tooth towards the buccal or palatal side, forward (mesioversion) or backward (distortion) |
| Inversion | Inverted tooth position (root versus alveolar ridge and crown versus basal bone portion). It is constantly accompanied by inclusion. | |
| Rotation | The element rotates along its longitudinal axis | |
| Intrusion | The tooth crown is on a lower plane than the occlusal plane | |
| Extrusion | The coronal margin is located higher than the occlusal plane | |
| Volume anomalies | Gigantism | Macrodontia |
| Dwarfism | Microdontia | |
| Taurodontism | Tooth with wide crown, short roots, extended pulp chamber | |
| Shape anomalies | Crown anomalies | Accessory cusps, conical or tuberculate shape of the crown |
| Roots anomalies | Anomalies in number, shape and size | |
| Endodontic anomalies | Anomalies in number, shape and size of the root canal morphology | |
| Developmental anomalies | Enamel pearl | Small hard rounded excrescence, located near the enamel-cement junction. |
| Fusion | Fusion of two teeth. Coalescence anomaly: due to close contact between germs; it can affect crowns and roots or only the crowns | |
| Concrescence | Two teeth are joined along the root surfaces by cementum. Coalescence anomaly | |
| Invaginated tooth | It is due to an infolding of enamel into dentine. There are two forms, coronal and radicular, with the coronal one being more common. | |
| Structural anomalies | Enamel anomalies | Quantitative and/or qualitative enamel defect affecting some or all teeth. Amelogenesis imperfecta: abnormal formation of the enamel, unrelated to any systemic or generalized conditions. Autosomal dominant or autosomal recessive or x-linked pattern. It affects both deciduous and permanent. |
| Dentin anomalies | Dentinogenesis imperfecta: brown teeth, crowns shrunk due to enamel flaking not supported by intact dentin. Autosomal dominant. It affects both deciduous and permanent. |
| Dento-Oro-Maxillofacial Structures Involved | Syndrome |
|---|---|
| Maxillary bones | Basal cell nevus syndrome (Gorlin syndrome) |
| Craniometaphyseal dysplasia | |
| Crouzon syndrome | |
| Fibrodysplasia Ossificans Progressiva | |
| Hyperparathyroidism-Jaw Tumor | |
| Mandibuloacral dysplasia with type A lipodystrophy | |
| Nager acrofacial dysostosis | |
| Congenital nemaline myopathies | |
| Osteopetrosis | |
| Maxillary bones and teeth | Ablepharon syndrome |
| Alagille syndrome | |
| Angelman syndrome | |
| Ankyloblepharon-ectodermal defects-cleft lip/palate (AEC) syndrome (type of Ectodermal Dysplasia) | |
| Cerebral, ocular, dental, auricular, skeletal anomalies (CODAS) syndrome | |
| CHARGE syndrome | |
| Cornelia de Lange syndrome | |
| Craniofrontonasal syndrome | |
| Dubowitz syndrome | |
| Ellis Van Creveld syndrome (Chondroectodermal dysplasia) | |
| Gardner syndrome | |
| Hallermann-Streiff syndrome | |
| Hutchinson-Gilford Progeria syndrome | |
| Hypophosphatemic rickets | |
| Kabuki syndrome (Niikawa–Kuroki syndrome) | |
| KBG syndrome | |
| Lenz microphthalmia | |
| Marfan syndrome | |
| Oculodento-osseous dysplasia | |
| Osteoglophonic dysplasia | |
| Progeroid syndrome | |
| Robinow syndrome | |
| Rubinstein Taydi syndrome | |
| Sanjad-Sakati syndrome | |
| Seckel syndrome | |
| Trichorhinophalangeal syndrome | |
| Maxillary bones and tongue | Fraser syndrome |
| Maxillary bones, teeth and soft tissue | Floating-Harbor syndrome |
| Larsen syndrome | |
| Lowe syndrome | |
| Rothmund-Thomson syndrome | |
| Silver Russel syndrome | |
| Maxillary bones, teeth and tongue | Beckwith-Wiedemann syndrome |
| Williams-Beuren syndrome (William syndrome) | |
| Soft tissue | Gingival fibromatosis with hypertrichosis syndrome |
| Kindler syndrome (type of Epidermolysis Bullosa) | |
| Pseudoxanthoma elasticum | |
| Recessive dystrophic epidermolysis bullosa of Hallopeau-Siemens | |
| Vascular Ehlers-Danlos syndrome | |
| Soft tissue and maxillary bones | Zimmermann-Laband-1 syndrome |
| Soft tissue and teeth | Aagenaes syndrome/lymphedema cholestasis syndrome |
| Prader-Willi Syndrome | |
| Rutherfurd syndrome | |
| Papillon-Lefèvre syndrome (type of Ectodermal Dysplasia) | |
| Soft tissue and tongue | Hyalinosis cutis et mucosae |
| Teeth | Amelogenesis imperfecta |
| Axenfeld-Rieger syndrome | |
| Dentin dysplasia | |
| Ectodermal dysplasia | |
| Focal dermal hypoplasia (Goltz syndrome) | |
| Jalili syndrome | |
| Oculo-facio-cardio-dental (OFCD) syndrome | |
| Witkop Tooth and Nail syndrome |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Salerno, C.; D’Avola, V.; Oberti, L.; Almonte, E.; Bazzini, E.M.; Tartaglia, G.M.; Cagetti, M.G. Rare Genetic Syndromes and Oral Anomalies: A Review of the Literature and Case Series with a New Classification Proposal. Children 2022, 9, 12. https://doi.org/10.3390/children9010012
Salerno C, D’Avola V, Oberti L, Almonte E, Bazzini EM, Tartaglia GM, Cagetti MG. Rare Genetic Syndromes and Oral Anomalies: A Review of the Literature and Case Series with a New Classification Proposal. Children. 2022; 9(1):12. https://doi.org/10.3390/children9010012
Chicago/Turabian StyleSalerno, Claudia, Valeria D’Avola, Luca Oberti, Elena Almonte, Elena Maria Bazzini, Gianluca Martino Tartaglia, and Maria Grazia Cagetti. 2022. "Rare Genetic Syndromes and Oral Anomalies: A Review of the Literature and Case Series with a New Classification Proposal" Children 9, no. 1: 12. https://doi.org/10.3390/children9010012