
Differential Diagnosis of Acquired and Hereditary Neuropathies in Children and Adolescents—Consensus-Based Practice Guidelines

 , , , , , , ,
, , , , , , ,
Abstract
:1. Introduction
2. Materials and Methods
- Strong recommendation: we recommend doing/recommend not doing
- Moderate recommendation: we suggest doing/suggest not doing
- Recommendation is open: may/can be done
- Strong consensus: >95% agreement of voters
- Consensus: >75–95% of voters
- Consent of majority: >50–75% of voters
- No consent of majority: <50% of voters
3. Definition and Classification of Neuropathies in Children and Adolescents
4. The Diagnostic Methods
4.1. History and Clinical Evaluation
4.2. Electrophysiological Diagnostics
- Evidence or exclusion of peripheral nerve damage
- Identification of the affected structures (motor, sensory or sensorimotor neuropathy)
- Determination of the pathomechanism (axonal, demyelinating, or mixed damage)
- Any signs of florid denervation or re-innervation
- Signs of any additional involvement of CNS structures
4.3. Sensory and Vegetative Functional Diagnostics
4.4. Imaging Diagnostics
4.5. Laboratory and Other Paraclinical Diagnostics
4.6. Neuropathological Diagnostics
4.7. Genetic Diagnostics
5. Differential Diagnosis of Acquired and Hereditary Neuropathies in Children and Adolescents
5.1. Nerve Injuries
5.2. Mononeuritis, Mononeuritis Multiplex
5.3. Guillain–Barré Syndrome (GBS) and Chronic Inflammatory Demyelinating Polyneuropathy (CIDP)
5.4. Toxic Neuropathies
5.5. Hereditary Non-Syndromic Neuropathies in Children and Adolescents
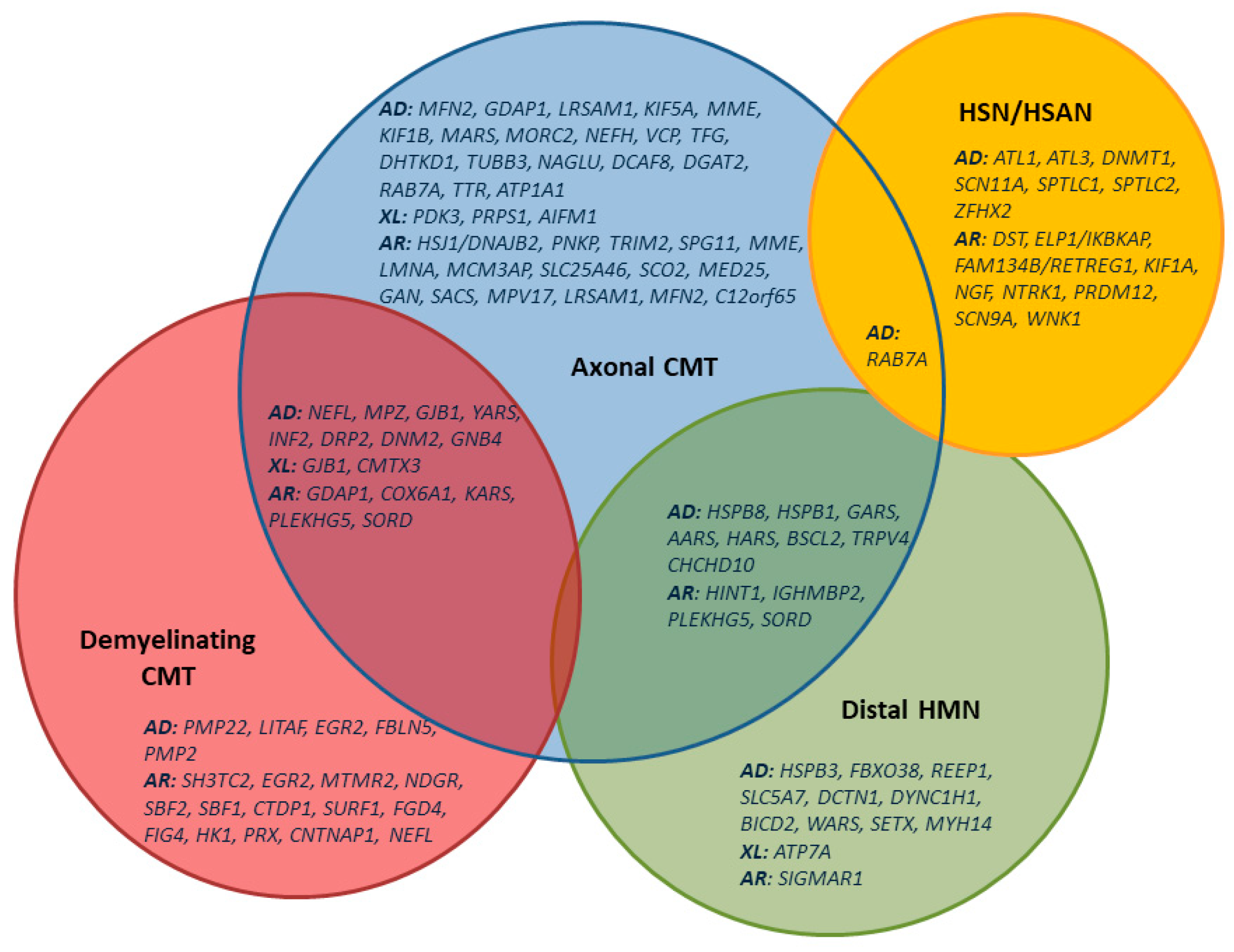
5.5.1. Hereditary Motor-Sensory Neuropathies (HMSN), Charcot–Marie–Tooth (CMT)-Neuropathy
5.5.2. Distal Hereditary Motor Neuropathies (dHMN)
5.5.3. Hereditary Sensory and Autonomous Neuropathies (HSAN, HSN)
5.5.4. Genetic Diagnostic Algorithms
5.6. Peripheral Neuropathies Associated with Complex Genetic Diseases
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Arbeitsgemeinschaft der Wissenschaftlichen Medizinischen Fachgesellschaften (AWMF)–Ständige Kommission Leitlinien. AWMF-Regelwerk Leitlinien. 1. Auflage. 2012. Available online: http://www.awmf.org/leitlinien/awmf-regelwerk.html (accessed on 20 December 2020).
- Papazian, O.; Alfonso, I.; Yaylali, I.; Velez, I.; Jayakar, P. Neurophysiological evaluation of children with traumatic radiculopathy, plexopathy, and peripheral neuropathy. Semin. Pediatr. Neurol. 2000, 7, 26–35. [Google Scholar] [CrossRef]
- Blankenburg, M.; Kraemer, N.; Hirschfeld, G.; Krumova, E.K.; Maier, C.; Hechler, T.; Aksu, F.; Magerl, W.; Reinehr, T.; Wiesel, T.; et al. Childhood diabetic neuropathy: Functional impairment and non-invasive screening assessment. Diabet. Med. 2012, 29, 1425–1432. [Google Scholar] [CrossRef]
- Louraki, M.; Karayianni, C.; Kanaka-Gantenbein, C.; Katsalouli, M.; Karavanaki, K. Peripheral neuropathy in children with type 1 diabetes. Diabet. Metab. 2012, 38, 281–289. [Google Scholar] [CrossRef]
- Royden Jones, H., Jr.; De Vivo, D.C.; Darras, B.T. (Eds.) Neuromuscular Disorders of Infancy, Childhood, and Adolescence; Butterworth: Amsterdam, The Netherlands, 2003. [Google Scholar]
- Williams, S.; Horrocks, I.A.; Ouvrier, R.A.; Gillis, J.; Ryan, M.M. Critical illness polyneuropathy and myopathy in pediatric intensive care: A review. Pediatr. Crit. Care Med. 2007, 8, 18–22. [Google Scholar] [CrossRef]
- Gilchrist, L. Chemotherapy-induced peripheral neuropathy in pediatric cancer patients. Semin. Pediatr. Neurol. 2012, 19, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Rudnik-Schöneborn, S.; Auer-Grumbach, M.; Senderek, J. Charcot-Marie-Tooth disease and hereditary motor neuropathies–update 2020. Medgen 2020, 32, 207–219. [Google Scholar]
- Yiu, E.M.; Ryan, M.M. Demyelinating prenatal and infantile developmental neuropathies. J. Peripher. Nerv. Syst. 2012, 17, 32–52. [Google Scholar] [CrossRef]
- Yiu, E.M.; Ryan, M.M. Genetic axonal neuropathies and neuronopathies of pre-natal and infantile onset. J. Peripher. Nerv. Syst. 2012, 17, 285–300. [Google Scholar] [CrossRef] [PubMed]
- Rossor, A.M.; Carr, A.S.; Devine, H.; Chandrashekar, H.; Pelayo-Negro, A.L.; Pareyson, D.; Shy, M.E.; Scherer, S.S.; Reilly, M.M. Peripheral neuropathy in complex inherited diseases: An approach to diagnosis. J. Neurol. Neurosurg. Psychiatry 2017, 88, 846–863. [Google Scholar] [CrossRef] [PubMed]
- Swoboda, K.J.; Edelbol-Eeg-Oloffson, K.; Harmon, R.L.; Bolton, C.F.; Harper, C.M.; Pitt, M.; Darras, B.T.; Royden Jones, H., Jr. Pediatric Electromyography. In Neuromuscular Disorders of Infancy, Childhood, and Adolescence; Royden Jones, H., Jr., De Vivo, D.C., Darras, B.T., Eds.; Butterworth: Amsterdam, The Netherlands, 2003. [Google Scholar]
- Pitt, M. Paediatric electromyography in the modern world: A personal view. Dev. Med. Child Neurol. 2011, 53, 120–124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pitt, M.C. Nerve conduction studies and needle EMG in very small children. Eur. J. Paediatr. Neurol. 2012, 16, 285–291. [Google Scholar] [CrossRef] [PubMed]
- Parano, E.; Uncini, A.; De Vivo, D.C.; Lovelace, R.E. Electrophysiologic correlates of peripheral nervous system maturation in infancy and childhood. J. Child. Neurol. 1993, 8, 336–338. [Google Scholar] [CrossRef]
- Plante-Bordeneuve, V. Transthyretin familial amyloid polyneuropathy: An update. J. Neurol. 2018, 265, 976–983. [Google Scholar] [CrossRef] [PubMed]
- Oaklander, A.L.; Nolano, M. Scientific Advances in and Clinical Approaches to Small-Fiber Polyneuropathy: A Review. JAMA Neurol. 2019, 76, 1240–1251. [Google Scholar] [CrossRef] [PubMed]
- Samadi, M.; Kazemi, B.; Golzari Oskoui, S.; Barzegar, M. Assessment of autonomic dysfunction in childhood Guillain-barré syndrome. J. Cardiovasc. Thorac. Res. 2013, 5, 81–85. [Google Scholar] [PubMed]
- Mulkey, S.B.; Glasier, C.M.; El-Nabbout, B.; Walters, W.D.; Ionita, C.; McCarthy, M.H.; Sharp, G.B.; Shbarou, R.M. Nerve root enhancement on spinal MRI in pediatric Guillain-Barre syndrome. Pediatr. Neurol. 2010, 43, 263–269. [Google Scholar] [CrossRef]
- Yikilmaz, A.; Doganay, S.; Gumus, H.; Per, H.; Kumandas, S.; Coskun, A. Magnetic resonance imaging of childhood Guillain-Barre syndrome. Childs Nerv. Syst. 2010, 26, 1103–1108. [Google Scholar] [CrossRef]
- Zuccoli, G.; Panigrahy, A.; Bailey, A.; Fitz, C. Redefining the Guillain-Barre spectrum in children: Neuroimaging findings of cranial nerve involvement. Am. J. Neuroradiol. 2011, 32, 639–642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pham, M.; Bäumer, T.; Bendszus, M. Peripheral nerves and plexus: Imaging by MR-neurography and high-resolution ultrasound. Curr. Opin. Neurol. 2014, 27, 370–379. [Google Scholar] [CrossRef]
- Grimm, A.; Décard, B.F.; Schramm, A.; Pröbstel, A.-K.; Rasenack, M.; Axer, H.; Fuhr, P. Ultrasound and electrophysiologic findings in patients with Guillain–Barré syndrome at disease onset and over a period of six months. Clin. Neurophysiol. 2016, 127, 1657–1663. [Google Scholar] [CrossRef] [PubMed]
- Berciano, J.; Sedano, M.J.; Pelayo-Negro, A.L.; García, A.; Orizaola, P.; Gallardo, E.; Lafarga, M.; Berciano, M.T.; Jacobs, B.C. Proximal nerve lesions in early Guillain–Barré syndrome: Implications for pathogenesis and disease classification. J. Neurol. 2017, 264, 221–236. [Google Scholar] [CrossRef] [PubMed]
- Eggermann, K.; Gess, B.; Häusler, M.; Weis, J.; Hahn, A.; Kurth, I. Hereditary Neuropathies. Dtsch. Arztebl. Int. 2018, 115, 91–97. [Google Scholar] [CrossRef] [PubMed]
- Katona, I.; Weis, J. Diseases of the peripheral nerves. Handb. Clin. Neurol. 2018, 145, 453–474. [Google Scholar]
- Weis, J.; Brandner, S.; Lammens, M.; Sommer, C.; Vallat, J.M. Processing of nerve biopsies: A practical guide for neuropathologists. Clin. Neuropathol. 2012, 31, 7–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weis, J.; Claeys, K.G.; Roos, A.; Azzedine, H.; Katona, I.; Schröder, J.M.; Senderek, J. Towards a functional pathology of hereditary neuropathies. Acta Neuropathol. 2017, 133, 493–515. [Google Scholar] [CrossRef]
- Nolano, M.; Tozza, S.; Caporaso, G.; Provitera, V. Contribution of Skin Biopsy in Peripheral neuropathies. Brain Sci. 2020, 10, 989. [Google Scholar] [CrossRef] [PubMed]
- Rauer, S.; Kastenbauer, S. Neuroborreliose, S3-Leitlinie 2018. In Deutsche Gesellschaft für Neurologie (Hrsg.). Leitlinien für Diagnostik und Therapie in der Neurologie. Available online: https://dgn.org/leitlinien/ll-030-071-2018-neuroborreliose/ (accessed on 28 June 2021).
- Malik, V.; Joshi, V.; Green, K.M.; Bruce, I.A. 15 minute consultation: A structured approach to the management of facial paralysis in a child. Arch. Dis. Child Educ. Pract. Ed. 2012, 97, 82–85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryan, M.M.; Tilton, A.; De Girolami, U.; Darras, B.T.; Jones, H.R., Jr. Paediatric mononeuritis multiplex: A report of three cases and review of the literature. Neuromuscul. Disord. 2003, 13, 751–756. [Google Scholar] [CrossRef]
- Garzoni, L.; Vanoni, F.; Rizzi, M.; Simonetti, G.D.; Goeggel Simonetti, B.; Ramelli, G.P.; Bianchetti, M.G. Nervous system dysfunction in Henoch-Schönlein syndrome: Systematic review of the literature. Rheumatology 2009, 48, 1524–1529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korinthenberg, R.; Trollmann, R.; Felderhoff-Müser, U.; Bernert, G.; Hackenberg, A.; Hufnagel, M.; Pohl, M.; Hahn, G.; Mentzel, H.J.; Sommer, C.; et al. Diagnosis and treatment of Guillain-Barré Syndrome in childhood and adolescence: An evidence- and consensus-based guideline. Eur. J. Paediatr. Neurol. 2020, 25, 5–16. [Google Scholar] [CrossRef] [PubMed]
- Nevo, Y.; Pestronk, A.; Kornberg, A.J.; Connolly, A.M.; Yee, W.C.; Iqbal, I.; Shield, L.K. Childhood chronic inflammatory demyelinating neuropathies: Clinical course and long-term follow-up. Neurology 1996, 47, 98–102. [Google Scholar] [CrossRef] [PubMed]
- McMillan, H.J.; Kang, P.B.; Jones, H.R.; Darras, B.T. Childhood chronic inflammatory demyelinating polyradiculoneuropathy: Combined analysis of a large cohort and eleven published series. Neuromuscul. Disord. 2013, 23, 103–111. [Google Scholar] [CrossRef]
- Korinthenberg, R. Chronic inflammatory demyelinating polyradiculoneuropathy in children and their response to treatment. Neuropediatrics 1999, 30, 190–196. [Google Scholar] [CrossRef] [PubMed]
- Devaux, J.J.; Miura, Y.; Fukami, Y.; Inoue, T.; Manso, C.; Belghazi, M.; Sekiguchi, K.; Kokubun, N.; Ichikawa, H.; Wong, A.H.; et al. Neurofascin-155 IgG4 in chronic inflammatory demyelinating polyneuropathy. Neurology 2016, 86, 800–807. [Google Scholar] [CrossRef] [Green Version]
- Vural, A.; Doppler, K.; Meinl, E. Autoantibodies Against the Node of Ranvier in Seropositive Chronic Inflammatory Demyelinating Polyneuropathy: Diagnostic, Pathogenic, and Therapeutic Relevance. Front. Immunol. 2018, 9, 1029. [Google Scholar] [CrossRef] [Green Version]
- De Simoni, D.; Ricken, G.; Winklehner, M.; Koneczny, I.; Karenfort, M.; Hustedt, U.; Seidel, U.; Abdel-Mannan, O.; Munot, P.; Rinaldi, S.; et al. Antibodies to nodal/paranodal proteins in paediatric immune-mediated neuropathy. Neurol. Neuroimmunol. Neuroinflamm. 2020, 7, e763. [Google Scholar] [CrossRef]
- Fernandez-Garcia, M.A.; Stettner, G.M.; Kinali, M.; Clarke, A.; Fallon, P.; Knirsch, U.; Wraige, E.; Jungbluth, H. Genetic neuropathies presenting with CIDP-like features in childhood. Neuromuscul. Disord. 2021, 31, 113–122. [Google Scholar] [CrossRef] [PubMed]
- Wilmshurst, J.M.; Ouvrier, R. Hereditary peripheral neuropathies of childhood: An overview for clinicians. Neuromuscul. Disord. 2011, 21, 763–775. [Google Scholar] [CrossRef] [PubMed]
- Pipis, M.; Rossor, A.M.; Laura, M.; Reilly, M.M. Next-generation sequencing in Charcot-Marie-Tooth disease: Opportunities and challenges. Nat. Rev. Neurol. 2019, 15, 644–656. [Google Scholar] [CrossRef]
- Baets, J.; Deconinck, T.; De Vriendt, E.; Zimoń, M.; Yperzeele, L.; Van Hoorenbeeck, K.; Peeters, K.; Spiegel, R.; Parman, Y.; Ceulemans, B.; et al. Genetic spectrum of hereditary neuropathies with onset in the first year of life. Brain 2011, 134, 2664–2676. [Google Scholar] [CrossRef] [PubMed]
- Sivera, R.; Sevilla, T.; Vílchez, J.J.; Martínez-Rubio, D.; Chumillas, M.J.; Vázquez, J.F.; Muelas, N.; Bataller, L.; Millán, J.M.; Palau, F.; et al. Charcot-Marie-Tooth disease. Genetic and clinical spectrum in a Spanish clinical series. Neurology 2013, 81, 1617–1625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manganelli, F.; Tozza, S.; Pisciotta, C.; Bellone, E.; Iodice, R.; Nolano, M.; Geroldi, A.; Capponi, S.; Mandich, P.; Santoro, L. Charcot-Marie-Tooth disease: Frequency of genetic subtypes in a Southern Italy population. J. Peripher. Nerv. Syst. 2014, 19, 292–298. [Google Scholar] [CrossRef]
- Rudnik-Schöneborn, S.; Tölle, D.; Senderek, J.; Eggermann, K.; Elbracht, M.; Kornak, U.; von der Hagen, U.; Kirschner, J.; Leube, B.; Müller-Felber, W.; et al. Diagnostic algorithms in Charcot-Marie-Tooth neuropathies: Experiences from a German genetic laboratory on the basis of 1206 index patients. Clin. Genet. 2016, 89, 34–43. [Google Scholar] [CrossRef]
- Liu, X.; Duan, X.; Zhang, Y.; Sun, A.; Fan, D. Molecular analysis and clinical diversity of distal hereditary motor neuropathy. Eur. J. Neurol. 2020, 27, 1319–1326. [Google Scholar] [CrossRef]
- Auer-Grumbach, M. Hereditary sensory and autonomic neuropathies. Handb. Clin. Neurol. 2013, 115, 893–906. [Google Scholar] [PubMed]
- Cox, J.J.; Woods, C.G.; Kurth, I. Peripheral sensory neuropathies–pain loss vs. pain gain. Medgen 2020, 32, 233–241. [Google Scholar]



{kind=link}
{kind=link}
| Hereditary Neuropathies | Acquired Neuropathies |
|---|---|
CMT2 (axonal, AD or AR) Intermediate CMT (AD or AR) CMT4 (demyelinating, AR) CMTX (demyelinating, axonal, XD, XR)
HSAN2-8 (AR)
DSMA (AR) DSMAX (XD)
HNA (AD)
Neuropathies in the context of neurodegenerative diseases |
Toxic (drugs, heavy metals) Metabolic/malnutrition (uremia, diabetes, dysproteinemia, vitamin deficiency, hypervitaminoses) Paraneoplastic Critical-illness neuropathy Small-fibre neuropathy
Traumatic Nerve tumours Nerve compression syndromes
Inflammatory |
| Motor Neurography | EMG | |
|---|---|---|
| Demyelination | Slowing of NCV, prolongation of distal-motor latency, prolongated or deficient F-waves (in proximal demyelination), abnormal dispersion of CMAP and partial conduction block (acquired multifocal demyelination, for example CIDP) | Normal findings when axonal anomalies are absent |
| Axonal damage | Lowered CMAP amplitude (can also be due to muscular atrophy/myopathy) | Florid denervation: fibrillation potentials, positive sharp waves, reduced interference pattern Chronic denervation with re-innervation: abnormal high-amplitude and polyphasic potentials, reduced interference pattern |
| Disorder | Lab Diagnostics |
|---|---|
| Infectious neuropathy | Serology and possible swabs, Borrelia burgdorferi, VZV, etc. |
| Inflammatory systemic diseases | ESR, CRP, ANA, double-stranded DNS-ab, C3 complement, ACE, lysozymes, immunoelectrophoretis, cryoglobulin |
| GBS, CIDP | CSF protein, CSF cell count, pathogen serology (CMV, Mycopl. pneumoniae, Campylobacter jejuni), anti-gangliosid antibodies (seldom positive), antibodies against paranodal proteins (neurofascin, etc.) |
| M. Refsum and other peroxisomopathies | Phytanic acid, VLCFA in serum |
| Bassen–Kornzweig syndrome (a-betalipoproteinemia) | Electrophoresis, lipid electrophoresis, vitamin E |
| Primary vitamin E resorption disorder | Vitamin E |
| Secondary vitamin resorption disorders | Vitamin D (Ca, P, AP, parathormone), vitamin E, vitamin K (INR, quick value), folic acid, vitamin B12 (with holotranscobalamin and methylmalonic acid), thiamin, riboflavin. Vitamin B6 (overdose?) |
| CDG syndromes | Isoelectric transferrin focussing |
| Metachromatic leukodystrophy and M. Krabbe | CSF protein, lysosomal enzymes |
| Mitochondriopathies | Lactate in plasma and CSF, possible muscle biopsy for biochemical analyses |
| Site of Lesion | Motor Defect | Sensory Defect | Aetiologies |
|---|---|---|---|
| Upper brachial plexus | Shoulder abduction and external rotation, elbow flexion, supination | Lateral/radial arm from shoulder to metacarpo-phalangeal joint of thumb | Strain/tearing of the shoulder, perinatal trauma, neuralgic amyotrophy, serogenetic neuritis, tumour infiltration |
| Lower brachial plexus | Finger and wrist flexion, finger ab- and -adduction, possibly Horner syndrome | Axillary and ulnar side of the arm from elbow to hand and finger IV and V | Trauma as above, abnormal cervical rib, scalenus syndrome, tumour infiltration |
| Long thoracic nerve | Elevation and rotation of scapula, winged scapula on shoulder flexion | - | »back packer palsy«, neuralgic amyotrophy |
| Radial nerve | Extension of wrist and metacarpo-phalangeal joints, abduction of thumb, extension of thumb and finger II (»flaccid drop of the hand«) | Back of the hand overlying metarcapals I and II | Humerus shaft fracture, pressure palsy |
| Median nerve | Flexion of wrist, flexion of finger I–III (»Schwurhand«) | Volar side of the hand and fingers I to the radial side of IV, dorsal side of same fingers | Supracondylar fracture of humerus, pressure palsy, carpal tunnel syndrome |
| Ulnar nerve | Flexion of wrist and metacarpo-phalangeal joint IV–V, ab-/adduction III–V, thumb adduction (»Krallenhand«) | Volar and dorsal side of hand and fingers IV and V (not radial side finger IV) | Supracondylar fracture of humerus, elbow fracture, pressure palsy |
| Ischiadic nerve | Combination of tibial- and peroneal lesion | Combination of tibial- and peroneal lesion | Ischiadic lesion at misplaced injection, pelvic fractures |
| Tibial nerve | Foot- and toe flexors, loss of Achilles tendon reflex | Plantar side of the foot, lateral side of the foot | Fractures, injury to the hollow of the knee |
| Peroneal nerve | Foot extension (»Steppage gait«) | Lateral lower leg, back of the foot | Fibular fracture, pressure palsy |
| Drugs | Heavy Metals and Solvents |
|---|---|
| Vincristin, cis-platinum | Lead |
| Taxane, epothilone | Gold |
| Bortezomib | Thallium |
| Thalidomide | Arsenic |
| Nitrofurantoin, isoniazid (INH) | Mercury |
| Hydantoine | n-Hexane |
| Chloramphenicol, metronidazol | Methyl-n-butylketone |
| Amphotericin | Triorthocresylphosphate |
| Disease | Diagnostics | Treatment |
|---|---|---|
| Refsum syndrome | Axonal or demyelinated NP, phytanic acid, pristanic acid | Phytanic acid-restricted diet, plasmapheresis |
| Adrenoleukodystrophy | VLCFA | Presymptomatic BMT |
| Metachromatic leukodystrophy | Demyelinated NP, lysosomal enzymes | Presymptomatic BMT |
| Vitamin E malabsorption | Vitamin E in serum | Vitamin E |
| Bassen–Kornzweig syndrome | Abetalipoproteinemia | Vitamin E |
| B12 deficiencies in resorption and utilisation | Vit B12 in serum, methylmalonic acis, homocystein in serum | Vitamin B12 |
| Folate deficiencies in resorption and utilisation | Folate, 5-Methyltetrahydrofolate | Folate, folinic acid |
| Cerebrotendinous xanthomatosis | Plasma cholestanol | Diet, chenodesoxycholic acid |
| Brown–Vialetto–van Laere syndrome | Bulbar paralysis, deafness, riboflavin in serum | Riboflavin |
| CD59 mutation | Anemia, paroxysmal nocturnal hematuria, relapsing NP | Eculizumab |
| Acute intermittent porphyria | Porphobilinogen | Glucose, haematin |
| Pyruvate dehydrogenase deficiency | Lactate, enzyme diagnostics, genetics | Ketogenic diet |
| Morbus Fabry (alpha-galactosidase A deficiency) | Severe acroparesthesias, autonomous neuropathy, angiokeratoma, corneal dystrophy, cardiovascular disease, stroke, renal impairment | Enzyme replacement therapy with agalsidase alfa or agalsidase beta |
| Familial amyloid polyneuropathy (onset from adolescence) | Symmetric sensory-motor and autonomic neuropathy, family history of neuropathy and/or cardiomyopathy, gi involvement, weight loss, carpal tunnel syndrome, TTR genetics | Liver transplantation, TTR stabilizers and gene modifying approaches in preparation [16] |
| Initially Presenting with PNP | +Ataxia | +Spasticity | +Ataxia +Spasticity +EPMS | +Global Developmental Delay | Multisystem Involvement |
|---|---|---|---|---|---|
| Metachromatic Leukodystrophy (ARSA) | Vitamin E deficiency (TTPA, MTP, acquired) * Vitamin B12 deficiency * Refsum syndrome (PHYA) * Sensory PNP in Friedreich Ataxia (FXN) EAOH (APTX) SCAR1(SETX) SCA27 (FGF14) AT (ATM) NARP (MTATP6) SCAR23 (PDYN) Microcephaly, seizures, and developmental delay (MCSZ) ARSACS (SACS) SCAN1 (TDP1) | Adrenoleukodystrophy (ABCD1) * Methylmalonic aciduria Vit. B12 deficiency * SPG (4, 9a, 12, 17, 39 ...) | Spastic Aataxia 5 (AFG3L2) LBSL (DARS2) Hypomyelinating leukodystrophy (TUBB4A) Leigh (-like) (SURF1/MFF, SUCLA2) | PDHC (PDHAl) * Krabbe (GALC) * Metachrom. Leukodystrophy (ARSA) * Aicardi-Goutieres syndrome * Global insensitivity to pain (CTLC1) Giant axonal neuropathy (GAN) NBIA2a (PLA2G6) CDG (NGLYl) MCSZ (PNKP) CEDNIK (SNAP29) Ponto-cerebellar hypoplasia (EXOSC3/AMPD2) Infantile Refsum (PEX7) | Mitochondriopathy * multi. acyl CoADH deficiency * Hexosaminidase A/B deficiency (HEXA/HEXB) * Brown–Vialetto–Van Laere (SLC52A2/3) * Peroxisomal 6 (PEX10) ACPHD (ABHD12) PHARC SPOE NBIA (C1901f12) SCAR21 (SCYLl) Familial Dystautonomia (TECPR2) Triple A (AAAS) MEDNIK (AP151) PTRH2 Galaktosialidosis (CTSA) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Korinthenberg, R.; Trollmann, R.; Plecko, B.; Stettner, G.M.; Blankenburg, M.; Weis, J.; Schoser, B.; Müller-Felber, W.; Lochbuehler, N.; Hahn, G.; et al. Differential Diagnosis of Acquired and Hereditary Neuropathies in Children and Adolescents—Consensus-Based Practice Guidelines. Children 2021, 8, 687. https://doi.org/10.3390/children8080687
Korinthenberg R, Trollmann R, Plecko B, Stettner GM, Blankenburg M, Weis J, Schoser B, Müller-Felber W, Lochbuehler N, Hahn G, et al. Differential Diagnosis of Acquired and Hereditary Neuropathies in Children and Adolescents—Consensus-Based Practice Guidelines. Children. 2021; 8(8):687. https://doi.org/10.3390/children8080687
Chicago/Turabian StyleKorinthenberg, Rudolf, Regina Trollmann, Barbara Plecko, Georg M. Stettner, Markus Blankenburg, Joachim Weis, Benedikt Schoser, Wolfgang Müller-Felber, Nina Lochbuehler, Gabriele Hahn, and et al. 2021. "Differential Diagnosis of Acquired and Hereditary Neuropathies in Children and Adolescents—Consensus-Based Practice Guidelines" Children 8, no. 8: 687. https://doi.org/10.3390/children8080687