
Additional Evidence for Neuropsychiatric Manifestations in Mosaic Trisomy 20: A Case Report and Brief Review

 ,
, 
Abstract
:1. Introduction
2. Case Report
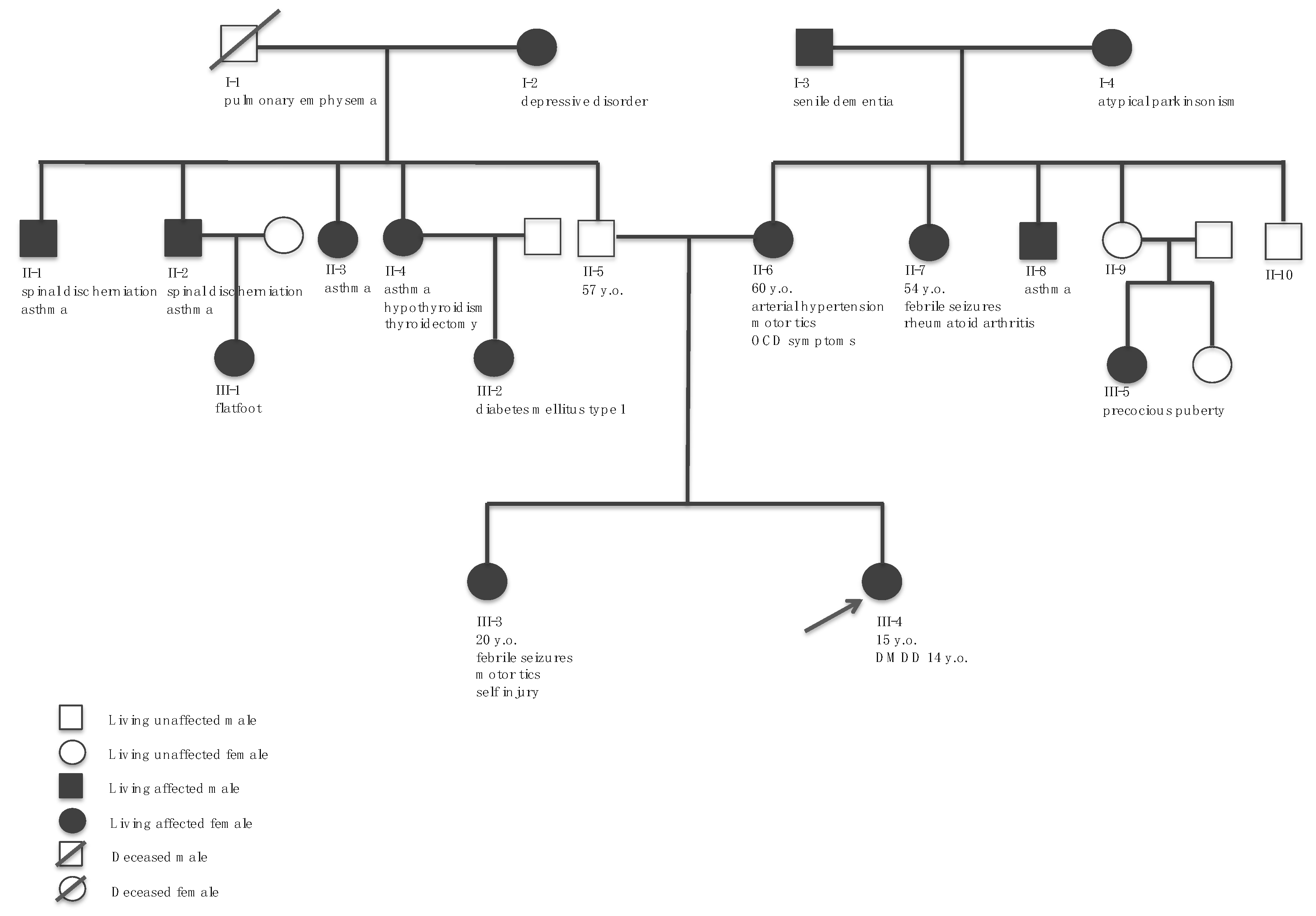
2.1. Medical History
2.2. Clinical Course, Diagnostic Conclusions, and Outpatient Follow-Up
3. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chuang, T.-H.; Chang, Y.-P.; Lee, M.-J.; Wang, H.-L.; Lai, H.-H.; Chen, S.-U. The Incidence of Mosaicism for Individual Chromosome in Human Blastocysts Is Correlated with Chromosome Length. Front. Genet. 2021, 11, 1677. [Google Scholar] [CrossRef]
- Pertile, M.D. Genome-Wide Cell-Free DNA-Based Prenatal Testing for Rare Autosomal Trisomies and Subchromosomal Abnormalities. In Noninvasive Prenatal Testing (NIPT); Page-Christiaens, L., Klein, H.-G., Eds.; Academic Press: Cambridge, MA, USA, 2018; pp. 97–123. [Google Scholar]
- Hsu, L.Y.F.; Kaffe, S.; Perlis, T.E. Trisomy 20 mosaicism in prenatal diagnosis–a review and update. Prenat. Diagn. 1987, 7, 581–596. [Google Scholar] [CrossRef] [PubMed]
- Hsu, L.Y.F.; Kaffe, S.; Perlis, T.E. A revisit of trisomy 20 mosaicism in prenatal diagnosis—An overview of 103 cases. Prenat. Diagn. 1991, 11, 7–15. [Google Scholar] [CrossRef] [PubMed]
- Montplaisir, R.; Lee, E.; Moreno-De-Luca, D.; Myers, W.C. Mosaic trisomy 20 and mitigation in capital crimes sentencing: A review and case report. Behav. Sci. Law 2019, 37, 512–521. [Google Scholar] [CrossRef] [PubMed]
- Korkontzelos, I. Prenatal diagnosis of trisomy 20 mosaicism associated with hypoplastic nasal bone as a single sonographic marker. Eur. J. Obstet. Gynecol. Reprod. Biol. 2017, 213, 140–141. [Google Scholar] [CrossRef]
- Velissariou, V.; Antoniadi, T.; Gyftodimou, J.; Bakou, K.; Grigoriadou, M.; Christopoulou, S.; Hatzipouliou, A.; Donoghue, J.; Karatzis, P.; Katsarou, E.; et al. Maternal uniparental isodisomy 20 in a foetus with trisomy 20 mosaicism: Clinical, cytogenetic and molecular analysis. Eur. J. Hum. Genet. 2002, 10, 694–698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mavromatidis, G.; Dinas, K.; Delkos, D.; Vosnakis, C.; Mamopoulos, A.; Rousso, D. Case of prenatally diagnosed non-mosaic trisomy 20 with minor abnormalities. J. Obstet. Gynaecol. Res. 2010, 36, 866–868. [Google Scholar] [CrossRef] [PubMed]
- Myers, T.L.; Prouty, L.A. Non-mosaic trisomy 20 in amniotic fluid cultures with minor anomalies in the fetus. Clin. Genet. 2008, 35, 233–236. [Google Scholar] [CrossRef]
- Ensenauer, R.E.; Shaughnessy, W.J.; Jalal, S.M.; Dawson, D.B.; Courteau, L.K.; Ellison, J.W. Trisomy 20 mosaicism caused by a maternal meiosis II error is associated with normal intellect but multiple congenital anomalies. Am. J. Med. Genet. Part A 2005, 134A, 202–206. [Google Scholar] [CrossRef]
- Warren, N.; Soukup, S.; King, J.; Dignan, P. Prenatal diagnosis of trisomy 20 by chorionic villus sampling (CVS): A case report with long-term outcome. Prenat. Diagn. 2001, 21, 1111–1113. [Google Scholar] [CrossRef]
- Strømme, P.; Van Der Hagen, C.B.; Haakonsen, M.; Risberg, K.; Hennekam, R. Follow-up of a girl with cleft lip and palate and multiple malformations: Trisomy 20 mosaicism. Scand. J. Plast. Reconstr. Surg. Hand Surg. 2005, 39, 178–179. [Google Scholar] [CrossRef] [PubMed]
- Reish, O.; Wolach, B.; Amiel, A.; Kedar, I.; Dolfin, T.; Fejgin, M. Dilemma of trisomy 20 mosaicism detected prenatally: Is it an innocent finding? Am. J. Med. Genet. 1998, 77, 72–75. [Google Scholar] [CrossRef]
- Powis, Z.; Erickson, R.P. Uniparental disomy and the phenotype of mosaic trisomy 20: A new case and review of the literature. J. Appl. Genet. 2009, 50, 293–296. [Google Scholar] [CrossRef] [PubMed]
- Girard, C.; Guillot, B.; Rivier, F.; Vale, F.D.; Bessis, D. Mosaïcisme pigmentaire de type Ito révélant une trisomie 20 en mosaïque. Ann. Dermatol. Venereol. 2005, 132, 151–153. [Google Scholar] [CrossRef]
- Morales, C.; Cuatrecasas, E.; Mademont-Soler, I.; Clusellas, N.; Peruga, E.; Català, V.; Garrido, C.; Milà, M.; Soler, A.; Sánchez, A. Non-mosaic trisomy 20 of paternal origin in chorionic villus and amniotic fluid also detected in fetal blood and other tissues. Eur. J. Med. Genet. 2010, 53, 197–200. [Google Scholar] [CrossRef] [PubMed]
- Karaoguz, M.Y.; Pala, E.; Kula, S.; Karaer, K.; Kan, D.; Nas, T.; Tunaoglu, S. Transposition of great arteries in an infant born after prenatal diagnosis of trisomy 20 mosaicism. Genet. Couns. 2007, 18, 437–443. [Google Scholar]
- Hartmann, A.; Hofmann, U.B.; Hoehn, H.; Broecker, E.B.; Hamm, H. Postnatal Confirmation of Prenatally Diagnosed Trisomy 20 Mosaicism in a Patient with Linear and Whorled Nevoid Hypermelanosis. Pediatr. Dermatol. 2004, 21, 636–641. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, C.C.; Hsu, J.J.; Lo, L.M.; Hsieh, T.T.; Soong, Y.K. Non-mosaic trisomy 20 in cultures of amniotic fluid from a fetus with serious congenital malformation. J. Formos. Med. Assoc. 1992, 91, 543–544. [Google Scholar]
- Willis, M.J.; Bird, L.M.; Dell’Aquilla, M.; Jones, M.C. Expanding the phenotype of mosaic trisomy 20. Am. J. Med. Genet. Part A 2008, 146A, 330–336. [Google Scholar] [CrossRef]
- Stein, Q.P.; Boyle, J.G.; Crotwell, P.L.; Flanagan, J.D.; Johnson, K.J.; Davis-Keppen, L.; Van Eerden, P.; Woltanski, A.R.; Watson, W.J. Prenatally diagnosed trisomy 20 mosaicism associated with arachnoid cyst of basal cistern. Prenat. Diagn. 2008, 28, 1169–1170. [Google Scholar] [CrossRef]
- Holzgreve, W.; Golabi, M.; Bradley, J. Multiple congenital anomalies in a child born after prenatal diagnosis of trisomy 20mosaicism. Clin. Genet. 1986, 29, 342–344. [Google Scholar] [CrossRef]
- Salafsky, I.S.; MacGregor, S.N.; Claussen, U.; Von Eggeling, F. Maternal UPD 20 in an infant from a pregnancy with mosaic trisomy 20. Prenat. Diagn. 2001, 21, 860–863. [Google Scholar] [CrossRef] [PubMed]
- Wallerstein, R.; Twersky, S.; Layman, P.; Kernaghan, L.; Aviv, H.; Pedro, H.F.; Pletcher, B. Long term follow-up of developmental delay in a child with prenatally-diagnosed trisomy 20 mosaicism. Am. J. Med. Genet. Part A 2005, 137A, 94–97. [Google Scholar] [CrossRef] [PubMed]
- Miny, P.; Karabacak, Z.; Hammer, P.; Schulte-Vallentin, M.; Holzgreve, W. Chromosome Analyses from Urinary Sediment: Postnatal Confirmation of a Prenatally Diagnosed Trisomy 20 Mosaicism. N. Engl. J. Med. 1989, 320, 809. [Google Scholar] [CrossRef] [PubMed]
- Achenbach, T.M.; Rescorla, L.A. Manual for the ASEBA School-Age Forms & Profiles; University of Vermont Research Center for Children, Youth & Families: Burlington, VT, USA, 2001. [Google Scholar]
- APA. Diagnostic and Statistical Manual of Mental Disorders, 5th ed.; American Psychiatric Publishing: Arlington, TX, USA, 2013. [Google Scholar]
- Wechsler, D. Wechsler Intelligence Scale for Children, 4th ed.; APA PsycNet: Washington, DC, USA, 2003. [Google Scholar]
- Ikeda, M.; Saito, T.; Kanazawa, T.; Iwata, N. Polygenic risk score as clinical utility in psychiatry: A clinical viewpoint. J. Hum. Genet. 2020, 66, 53–60. [Google Scholar] [CrossRef] [PubMed]
- Cimino, S.; Carola, V.; Cerniglia, L.; Bussone, S.; Bevilacqua, A.; Tambelli, R. The μ-opioid receptor gene A118G polymorphism is associated with insecure attachment in children with disruptive mood regulation disorder and their mothers. Brain Behav. 2020, 10, e01659. [Google Scholar] [CrossRef]
- Roberson-Nay, R.; Leibenluft, E.; Brotman, M.A.; Myers, J.; Larsson, H.; Lichtenstein, P.; Kendler, K.S. Longitudinal Stability of Genetic and Environmental Influences on Irritability: From Childhood to Young Adulthood. Am. J. Psychiatry 2015, 172, 657–664. [Google Scholar] [CrossRef] [Green Version]
- Moore, A.A.; Lapato, D.M.; Brotman, M.A.; Leibenluft, E.; Aggen, S.H.; Hettema, J.M.; York, T.P.; Silberg, J.L.; Roberson-Nay, R. Heritability, stability, and prevalence of tonic and phasic irritability as indicators of disruptive mood dysregulation disorder. J. Child Psychol. Psychiatry 2019, 60, 1032–1041. [Google Scholar] [CrossRef]
- Starr, L.R.; Dienes, K.; Li, Y.I.; Shaw, Z.A. Chronic stress exposure, diurnal cortisol slope, and implications for mood and fatigue: Moderation by multilocus HPA-Axis genetic variation. Psychoneuroendocrinology 2019, 100, 156–163. [Google Scholar] [CrossRef]
- Dougherty, L.R.; Barrios, C.S.; Carlson, G.A.; Klein, D.N. Predictors of Later Psychopathology in Young Children with Disruptive Mood Dysregulation Disorder. J. Child Adolesc. Psychopharmacol. 2017, 27, 396–402. [Google Scholar] [CrossRef]
- Arinami, T.; Ohtsuki, T.; Ishiguro, H.; Ujike, H.; Tanaka, Y.; Morita, Y.; Mineta, M.; Takeichi, M.; Yamada, S.; Imamura, A.; et al. Genomewide High-Density SNP Linkage Analysis of 236 Japanese Families Supports the Existence of Schizophrenia Susceptibility Loci on Chromosomes 1p, 14q, and 20p. Am. J. Hum. Genet. 2005, 77, 937–944. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Detera-Wadleigh, S.D.; Badner, J.A.; Yoshikawa, T.; Sanders, A.R.; Goldin, L.R.; Turner, G.; Rollins, D.Y.; Moses, T.; Guroff, J.J.; Kazuba, D.; et al. Initial Genome Scan of the NIMH Genetics Initiative Bipolar Pedigrees: Chromosomes 4, 7, 9, 18, 19, 20, and 21q. Am. J. Med. Genet. 1997, 74, 254–262. [Google Scholar] [CrossRef]
- Lewis, C.; Levinson, D.F.; Wise, L.H.; DeLisi, L.E.; Straub, R.E.; Hovatta, I.; Williams, N.M.; Schwab, S.G.; Pulver, A.E.; Faraone, S.; et al. Genome Scan Meta-Analysis of Schizophrenia and Bipolar Disorder, Part II: Schizophrenia. Am. J. Hum. Genet. 2003, 73, 34–48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moises, H.; Yang, L.; Kristbjarnarson, H.; Wiese, C.; Byerley, W.; Macciardi, F.; Arolt, V.; Blackwood, D.; Liu, X.; Sjögren, B.; et al. An international two–stage genome–wide search for schizophrenia susceptibility genes. Nat. Genet. 1995, 11, 321–324. [Google Scholar] [CrossRef]
- Ross, J.; Berrettini, W.; Coryell, W.; Gershon, E.S.; Badner, J.A.; Kelsoe, J.R.; McInnis, M.G.; McMahon, F.; Murphy, D.L.; Nurnberger, J.; et al. Genome-wide parametric linkage analyses of 644 bipolar pedigrees suggest susceptibility loci at chromosomes 16 and 20. Psychiatr. Genet. 2008, 18, 191–198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Radhakrishna, U.; Senol, S.; Herken, H.; Gücüyener, K.; Gehrig, C.; Blouin, J.-L.; Akarsu, N.; Antonarakis, S. An apparently dominant bipolar affective disorder (BPAD) locus on chromosome 20p11.2–q11.2 in a large Turkish pedigree. Eur. J. Hum. Genet. 2001, 9, 39–44. [Google Scholar] [CrossRef]
- Bigdeli, T.B.; Maher, B.S.; Zhao, Z.; Oord, E.J.C.G.V.D.; Thiselton, D.L.; Sun, J.; Webb, B.T.; Amdur, R.L.; Wormley, B.; O’Neill, F.A.; et al. Comprehensive Gene-Based Association Study of a Chromosome 20 Linked Region Implicates Novel Risk Loci for Depressive Symptoms in Psychotic Illness. PLoS ONE 2011, 6, e21440. [Google Scholar] [CrossRef]
- Hamshere, M.L.; Schulze, T.G.; Schumacher, J.; Corvin, A.; Owen, M.J.; Jamra, R.A.; Propping, P.; Maier, W.; Diaz, G.O.Y.; Mayoral, F.; et al. Mood-incongruent psychosis in bipolar disorder: Conditional linkage analysis shows genome-wide suggestive linkage at 1q32.3, 7p13 and 20q13.31. Bipolar Disord. 2009, 11, 610–620. [Google Scholar] [CrossRef]
- Erlangsen, A.; Appadurai, V.; Wang, Y.; Turecki, G.; Mors, O.; Werge, T.; Mortensen, P.B.; Starnawska, A.; Børglum, A.; Schork, A.; et al. Genetics of suicide attempts in individuals with and without mental disorders: A population-based genome-wide association study. Mol. Psychiatry 2018, 25, 2410–2421. [Google Scholar] [CrossRef]
- Li, M.; Luo, X.-J.; Landén, M.; Bergen, S.; Hultman, C.M.; Li, X.; Zhang, W.; Yao, Y.-G.; Zhang, C.; Liu, J.; et al. Impact of a cis-associated gene expression SNP on chromosome 20q11.22 on bipolar disorder susceptibility, hippocampal structure and cognitive performance. Br. J. Psychiatry 2016, 208, 128–137. [Google Scholar] [CrossRef] [Green Version]
- Peron, A.; Catusi, I.; Recalcati, M.P.; Calzari, L.; Larizza, L.; Vignoli, A.; Canevini, M.P. Ring Chromosome 20 Syndrome: Genetics, Clinical Characteristics, and Overlapping Phenotypes. Front. Neurol. 2020, 11, 1617. [Google Scholar] [CrossRef] [PubMed]
- Kamath, B.M.; Thiel, B.D.; Gai, X.; Conlin, L.K.; Munoz, P.S.; Glessner, J.; Clark, D.; Warthen, D.M.; Shaikh, T.H.; Mihci, E.; et al. SNP array mapping of chromosome 20p deletions: Genotypes, phenotypes, and copy number variation. Hum. Mutat. 2009, 30, 371–378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khattak, S.; Jan, M.; Warsi, S.; Khattak, S. Chromosome 20p Partial De Novo Duplication Identified in a Female Paediatric Patient with Characteristic Facial Dysmorphism and Behavioural Anomalies. Case Rep. Genet. 2020, 2020, 7093409. [Google Scholar] [CrossRef] [PubMed]
- Hanafusa, H.; Morisada, N.; Ishida, Y.; Sakata, R.; Morita, K.; Miura, S.; Ye, M.J.; Yamamoto, T.; Okamoto, N.; Nozu, K.; et al. The smallest de novo 20q11.2 microdeletion causing intellectual disability and dysmorphic features. Hum. Genome Var. 2017, 4, 17050. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Dijck, A.; Silfhout, A.T.V.-V.; Cappuyns, E.; van der Werf, I.M.; Mancini, G.M.; Tzschach, A.; Bernier, R.; Gozes, I.; Eichler, E.E.; Romano, C.; et al. Clinical Presentation of a Complex Neurodevelopmental Disorder Caused by Mutations in ADNP. Biol. Psychiatry 2018, 85, 287–297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Breen, M.S.; Garg, P.; Tang, L.; Mendonca, D.; Levy, T.; Barbosa, M.; Arnett, A.B.; Kurtz-Nelson, E.; Agolini, E.; Battaglia, A.; et al. Episignatures Stratifying Helsmoortel-Van Der Aa Syndrome Show Modest Correlation with Phenotype. Am. J. Hum. Genet. 2020, 107, 555–563. [Google Scholar] [CrossRef]
- Bagyinszky, E.; Van Giau, V.; Youn, Y.C.; An, S.S.A.; Kim, S. Characterization of mutations in PRNP (prion) gene and their possible roles in neurodegenerative diseases. Neuropsychiatr. Dis. Treat. 2018, 14, 2067–2085. [Google Scholar] [CrossRef] [Green Version]
- Griffith, C.B.; Vance, G.H.; Weaver, D.D. Phenotypic variability in trisomy 13 mosaicism: Two new patients and literature review. Am. J. Med. Genet. Part A 2009, 149A, 1346–1358. [Google Scholar] [CrossRef] [PubMed]
- Pillinger, T.; D’Ambrosio, E.; McCutcheon, R.; Howes, O.D. Is psychosis a multisystem disorder? A meta-review of central nervous system, immune, cardiometabolic, and endocrine alterations in first-episode psychosis and perspective on potential models. Mol. Psychiatry 2018, 24, 776–794. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
| Body Systems and Functions | Symptoms/Signs | Current Case + Present X Not Present |
|---|---|---|
| Craniofacial | ||
| Korkontzelos (2017) [6], Velissariou et al. (2002) [7] | Underdeveloped nasal bone | + |
| Hsu et al. (1991) [4], Mavromatidis et al. (2010) [8], Myers and Prouty (1989) [9], Ensenauer et al. (2005) [10] | Ear morphology abnormalities | + |
| Velissariou et al. (2002) [7], Warren et al. (2001) [11] | Micrognathia and retrognathia | X |
| Stromme et al. (2005) [12] | Cleft lip and palate | X |
| Hsu et al. (1991) [4], Velissariou et al. (2002) [7], Reish et al. (1998) [13] | Abnormal periorbital region morphology | X |
| Cutaneous | ||
| Powis and Erickson (2009) [14] | Thin and brittle nails | X |
| Warren et al. (2001) [11], Powis and Erickson (2009) [14], Girard et al. (2005) [15] | Hypomelanosis of Ito | X |
| Hartmann et al. (2004) [18] | Linear and whorled nevoid hypermelanosis | X |
| Velissariou et al. (2002) [7] | Mongolian spots | X |
| Cardiovascular-Pulmonary | ||
| Morales et al. (2010) [16] | Pulmonary isomerism | X |
| Velissariou et al. (2002) [7], Karaoguz et al. (2007) [17], Hartmann et al. (2004) [18], Hsieh et al. (1992) [19] | Congenital heart defects | X |
| Gastrointestinal | ||
| Willis et al. (2008) [20] | Stipsis | + |
| Endocrinological | ||
| Girard et al. (2005) [15] | Growth hormone deficiency | X |
| Reproductive | ||
| Ensenauer et al. (2005) [10], Girard et al. (2005) [15] | Cryptorchidism | X |
| Locomotor | ||
| Willis et al. (2008) [20], Stein et al. (2008) [21] | Sloped shoulders | + |
| Velissariou et al. (2002) [7], Willis et al. (2008) [20] | Abnormal spinal column morphology | + (Scoliosis) |
| Velissariou et al. (2002) [7], Reish et al. (1998) [13], Willis et al. (2008) [20], Stein et al. (2008) [21], Holzgreve et al. (1986) [22] | Central and peripheral altered muscle tone | + (Hypotonia) |
| Velissariou et al. (2002) [7] | Ligamentous laxity | + |
| Morales et al. (2010) [16] | Camptodactyly | X |
| Hsu et al. (1991) [4], Velissariou et al. (2002) [7] | Clinodactyly | X |
| Montplaisir (2019) [5] | Polydactyly | X |
| Stein et al. (2008) [21] | Rib anomalies | X |
| Nervous | ||
| Powis and Erickson (2009) [14] | Epileptic manifestations | X |
| Ensenauer et al. (2005) [10] | Hearing impairment | X |
| Hsu et al. (1991) [4], Salafsky et al. (2001) [23] | Microcephaly | X |
| Hsieh et al. (1992) [19], Stein et al. (2008) [21] | Abnormal neuroimaging findings | X |
| Cognitive and Mental | ||
| Willis et al. (2008) [20] | Preserved IQ and learning disabilities | + |
| Repetitive behaviors | + | |
| Isolation | + | |
| Social and emotional difficulties | + | |
| Pragmatic/Social communication difficulties | + | |
| Wallerstein et al. (2005) [24] | Atypical neurodevelopment with attention deficit | X |
| Impulsivity | + | |
| Atypical social reciprocity | + | |
| Girard et al. (2005) [15] | Speech difficulties | X |
| Hsu et al. (1991) [4], Velissariou et al. (2002) [7], Girard et al. (2005) [15], Hartmann et al. (2004) [18], Salafsky et al. (2001) [23], Miny et al. (1989) [25] | Developmental psychomotor difficulties | + |
| Reish et al. (1998) [13] | Impaired fine and gross motor abilities | + |
| Holzgreve et al. (1986) [22] | Developmental language difficulties | X |
| Montplaisir (2019) [5] | Self-injury | + |
| Montplaisir (2019) [5] | Hallucinations | X |
| Neurocognitive Assessment | |||
|---|---|---|---|
| Wechsler Intelligence Scale for Children–Fourth Edition (WISC-IV) | Score (95% Confidence Interval; subtest raw score) | ||
| Full-Scale Intelligence Quotient | 80 (75–87) | ||
| Verbal Comprehension | 78 (72–86; Similarities: 7; Comprehension: 3; Vocabulary: 7; Information (supplemental subtest): 5) | ||
| Perceptual Reasoning | 95 (87–103; Block Design: 11; Matrix Reasoning: 9; Picture Concepts: 8) | ||
| Working Memory | 79 (72–90; Subtest: Digit Span: 7; Letter–Number Sequencing: 6; Arithmetic (supplemental subtest): 3) | ||
| Processing Speed | 85 (77–97; Coding: 8; Symbol Search: 7) | ||
| Psychological Assessment | |||
| Child Behavior Checklist (CBCL) | T-Scores (Patient) | T-Scores (Mother) | Clinical: T ≥ 70 Borderline: 65 ≥ T < 70 Non-clinical: T < 65 |
| Syndrome Scale Scores | |||
| Social Problems | 70 | 70 | Clinical |
| Thought Problems | 73 | 73 | Clinical |
| Attention Problems | 83 | 70 | Clinical |
| Internalizing Problems | |||
| Withdrawn/Depressed | 100 | 100 | Clinical |
| Anxious/Depressed | 100 | 74 | Clinical |
| Somatic Complaints | 70 | 65 | Clinical/Borderline |
| Externalizing Problems | |||
| Rule-Breaking Behavior | 60 | 57 | Non-clinical |
| Aggressive Behavior | 58 | 58 | Non-clinical |
| Total Problems | |||
| Internalizing Score | 86 | 77 | Clinical |
| Externalizing Score | 59 | 58 | Non-clinical |
| Total Problems Score | 74 | 70 | Clinical |
| DSM-Oriented Scales | |||
| Depressive Problems | 88 | 79 | Clinical |
| Anxiety Problems | 94 | 73 | Clinical |
| Somatic Problems | 59 | 59 | Non-clinical |
| Attention Deficit | 69 | 60 | Borderline/Non-clinical |
| Oppositional Defiant Problems | 59 | 63 | Non-clinical |
| Conduct Problems | 61 | 55 | Non-clinical |
| Other Scales | |||
| Sluggish Cognitive Tempo | 80 | 73 | Clinical |
| Obsessive-Compulsive Problems | 87 | 66 | Clinical/Borderline |
| Stress Problems | 86 | 78 | Clinical |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Colizzi, M.; Antolini, G.; Passarella, L.; Rizzo, V.; Puttini, E.; Zoccante, L. Additional Evidence for Neuropsychiatric Manifestations in Mosaic Trisomy 20: A Case Report and Brief Review. Children 2021, 8, 1030. https://doi.org/10.3390/children8111030
Colizzi M, Antolini G, Passarella L, Rizzo V, Puttini E, Zoccante L. Additional Evidence for Neuropsychiatric Manifestations in Mosaic Trisomy 20: A Case Report and Brief Review. Children. 2021; 8(11):1030. https://doi.org/10.3390/children8111030
Chicago/Turabian StyleColizzi, Marco, Giulia Antolini, Laura Passarella, Valentina Rizzo, Elena Puttini, and Leonardo Zoccante. 2021. "Additional Evidence for Neuropsychiatric Manifestations in Mosaic Trisomy 20: A Case Report and Brief Review" Children 8, no. 11: 1030. https://doi.org/10.3390/children8111030