
Genetic Testing among Children in a Complex Care Program

Abstract
:1. Introduction
2. Methods
2.1. Setting
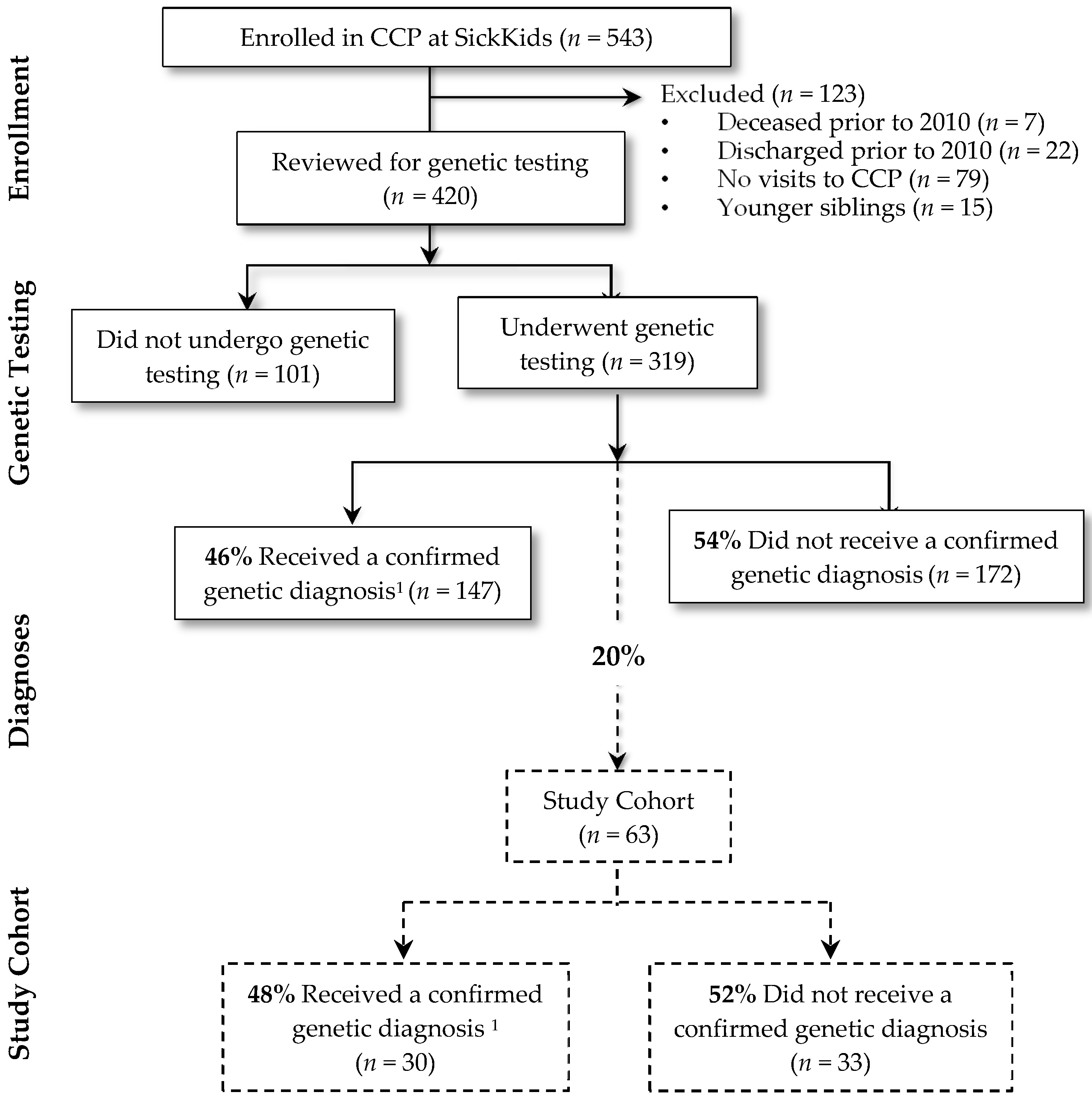
2.2. Study Cohort
2.3. Data Collection Procedures and Measures
2.3.1. Data Sources
2.3.2. Demographic and Clinical Characteristics
2.3.3. Genetic Tests
2.4. Costs
2.5. Analysis
3. Results
3.1. Characteristics of the Cohort (n = 63)
3.2. Genetic Tests
3.3. Length of Testing Period
3.4. Genetic Testing Costs
3.5. Types of Genetic Testing
4. Discussion
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Cohen, E.; Kuo, D.Z.; Agrawal, R.; Berry, J.G.; Bhagat, S.K.; Simon, T.D.; Srivastava, R. Children with medical complexity: An emerging population for clinical and research initiatives. Pediatrics 2011, 127, 529–538. [Google Scholar] [CrossRef] [PubMed]
- Burns, K.H.; Casey, P.H.; Lyle, R.E.; Bird, T.M.; Fussell, J.J.; Robbins, J.M. Increasing prevalence of medically complex children in US hospitals. Pediatrics 2010, 126, 638–646. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, R.; Antonelli, R.C. Hospital-based programs for children with special health care needs: Implications for health care reform. Arch. Pediatr. Adolesc. Med. 2011, 165, 570–572. [Google Scholar] [CrossRef] [PubMed]
- Dewan, T.; Cohen, E. Children with medical complexity in Canada. Paediatr. Child. Health 2013, 18, 518–522. [Google Scholar] [CrossRef] [PubMed]
- Berry, J.G.; Agrawal, R.; Kuo, D.Z.; Cohen, E.; Risko, W.; Hall, M.; Casey, P.; Gordon, J.; Srivastava, R. Characteristics of hospitalizations for patients who use a structured clinical care program for children with medical complexity. J. Pediatr. 2011, 159, 284–290. [Google Scholar] [CrossRef] [PubMed]
- Hilbert, J.E.; Ashizawa, T.; Day, J.W.; Luebbe, E.A.; Martens, W.B.; McDermott, M.P.; Tawil, R.; Thornton, C.A.; Moxley Iii, R.T. Diagnostic odyssey of patients with myotonic dystrophy. J. Neurol. 2013, 260, 2497–2504. [Google Scholar] [CrossRef] [PubMed]
- Wong, S.H.; McClaren, B.J.; Archibald, A.D.; Weeks, A.; Langmaid, T.; Ryan, M.M.; Kornberg, A.; Metcalfe, S.A. A mixed methods study of age at diagnosis and diagnostic odyssey for Duchenne muscular dystrophy. Eur. J. Hum. Genet. 2015, 23, 1294–1300. [Google Scholar] [CrossRef] [PubMed]
- Lawton, S.; Hickerton, C.; Archibald, A.D.; McClaren, B.J.; Metcalfe, S.A. A mixed methods exploration of families’ experiences of the diagnosis of childhood spinal muscular atrophy. J. Hum. Genet. 2015, 23, 575–580. [Google Scholar] [CrossRef] [PubMed]
- Knott, M.; Leonard, H.; Downs, J. The diagnostic odyssey to Rett syndrome: The experience of an Australian family. Am. J. Med. Genet. Part A 2012, 158, 10. [Google Scholar] [CrossRef] [PubMed]
- Mroch, A.R.; Flanagan, J.D.; Stein, Q.P. Solving the puzzle: Case examples of array comparative genomic hybridization as a tool to end the diagnostic odyssey. Curr. Probl. Pediatr. Adolesc. Health Care 2012, 42, 74–78. [Google Scholar] [CrossRef] [PubMed]
- Yuen, R.K.C.; Thiruvahindrapuram, B.; Merico, D.; Walker, S.; Tammimies, K.; Hoang, N.; Chrysler, C.; Nalpathamkalam, T.; Pellecchia, G.; Liu, Y. Whole-genome sequencing of quartet families with autism spectrum disorder. Nat. Med. 2015, 21, 185–191. [Google Scholar] [CrossRef]
- Yang, Y.; Muzny, D.M.; Xia, F.; Niu, Z.; Person, R.; Ding, Y.; Ward, P.; Braxton, A.; Wang, M.; Buhay, C. Molecular findings among patients referred for clinical whole-exome sequencing. JAMA 2014, 312, 1870–1879. [Google Scholar] [CrossRef] [PubMed]
- Stavropoulos, D.J.; Merico, D.; Jobling, R.; Bowdin, S.; Monfared, N.; Thiruvahindrapuram, B.; Nalpathamkalam, T.; Pellecchia, G.; Yuen, R.K.C.; Szego, M.J. Whole-genome sequencing expands diagnostic utility and improves clinical management in paediatric medicine. NPJ Genom. Med. 2016, 1, 15012. [Google Scholar] [CrossRef]
- Beale, S.; Sanderson, D.; Sanniti, A.; Dundar, Y.; Boland, A. A scoping study to explore the cost-effectiveness of next-generation sequencing compared with traditional genetic testing for the diagnosis of learning disabilities in children. Health Technol. Assess. 2015, 19, 1–90. [Google Scholar] [CrossRef] [PubMed]
- Bowdin, S.; Ray, P.N.; Cohn, R.D.; Meyn, M.S. The genome clinic: A multidisciplinary approach to assessing the opportunities and challenges of integrating genomic analysis into clinical care. Hum. Mutat. 2014, 35, 513–519. [Google Scholar] [CrossRef] [PubMed]
- Manolio, T.A.; Abramowicz, M.; Al-Mulla, F.; Anderson, W.; Balling, R.; Berger, A.C.; Bleyl, S.; Chakravarti, A.; Chantratita, W.; Chisholm, R.L.; et al. Global implementation of genomic medicine: We are not alone. Sci. Transl. Med. 2015, 7, 290ps213. [Google Scholar] [CrossRef] [PubMed]
- Bowdin, S.C.; Hayeems, R.Z.; Monfared, N.; Cohn, R.D.; Meyn, M.S. The Sickkids genome clinic: Developing and evaluating a pediatric model for individualized genomic medicine. Clin. Genet. 2016, 89, 10. [Google Scholar] [CrossRef] [PubMed]
- Cohen, E.; Berry, J.G.; Camacho, X.; Anderson, G.; Wodchis, W.; Guttmann, A. Patterns and costs of health care use of children with medical complexity. Pediatrics 2012, 130, e1463. [Google Scholar] [CrossRef] [PubMed]
- Adams, S.; Cohen, E.; Mahant, S.; Friedman, J.N.; MacCulloch, R.; Nicholas, D.B. Exploring the usefulness of comprehensive care plans for children with medical complexity (CMC): A qualitative study. BMC Pediatr. 2013, 13, 1. [Google Scholar] [CrossRef] [PubMed]
- Miller, D.T.; Adam, M.P.; Aradhya, S.; Biesecker, L.G.; Brothman, A.R.; Carter, N.P.; Church, D.M.; Crolla, J.A.; Eichler, E.E.; Epstein, C.J. Consensus statement: Chromosomal microarray is a first-tier clinical diagnostic test for individuals with developmental disabilities or congenital anomalies. Am. J. Hum. Genet. 2010, 86, 749–764. [Google Scholar] [CrossRef] [PubMed]
- Guide 1.4, Chapter 3.3. Eurocat Subgroups of Congenital Anomalies (Version 2014; Implemented in Edmp December 2014, Used for Website Prevalence Tables from December 2014). Available online: http://www.eurocat-network.eu/content/EUROCAT-Guide-1.4-Section-3.3.pdf (accessed on 23 February 2017).
- Annual Average Exchange Rates. Available online: http://www.bankofcanada.ca/rates/exchange/annual-average-exchange-rates/ (accessed on 23 February 2017).
- Consumer Price Index Cansim Table 326-0021. Available online: http://www5.statcan.gc.ca/cansim/a26?id=3260021 (accessed on 23 February 2017).
- Census Metropolitan Area of Toronto, Ontario. Available online: https://www12.statcan.gc.ca/census-recensement/2011/as-sa/fogs-spg/Facts-cma-eng.cfm?LANG=Eng&GK=CMA&GC=535 (accessed on 23 February 2017).
- Population of Census Metropolitan Areas. Available online: http://www.statcan.gc.ca/tables-tableaux/sum-som/l01/cst01/demo05a-eng.htm (accessed on 23 February 2017).
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders, 5th ed.; American Psychiatric Publishing: Arlington, VA, USA, 2013; ISBN 978-0890425558. [Google Scholar]
- Van Nimwegen, K.J.M.; Schieving, J.H.; Willemsen, M.; Veltman, J.A.; van der Burg, S.; van der Wilt, G.J.; Grutters, J.P.C. The diagnostic pathway in complex paediatric neurology: A cost analysis. Eur. J. Paediatr. Neurol. 2015, 19, 233–239. [Google Scholar] [CrossRef] [PubMed]
- Richards, J.; Korgenski, E.K.; Srivastava, R.; Bonkowsky, J.L. Costs of the diagnostic odyssey in children with inherited leukodystrophies. Neurology 2015, 85, 1167–1170. [Google Scholar] [CrossRef] [PubMed]
- Tsiplova, K.; Zur, R.M.; Ungar, W.J. A Microcosting and Cost-Consequence Analysis of Genomic Testing Strategies in Autism Spectrum Disorder; 2016-02.2; The Hospital for Sick Children, Technology Assessment at SickKids (TASK): Toronto, ON, Canada, 21 September 2016. [Google Scholar]



{kind=link}
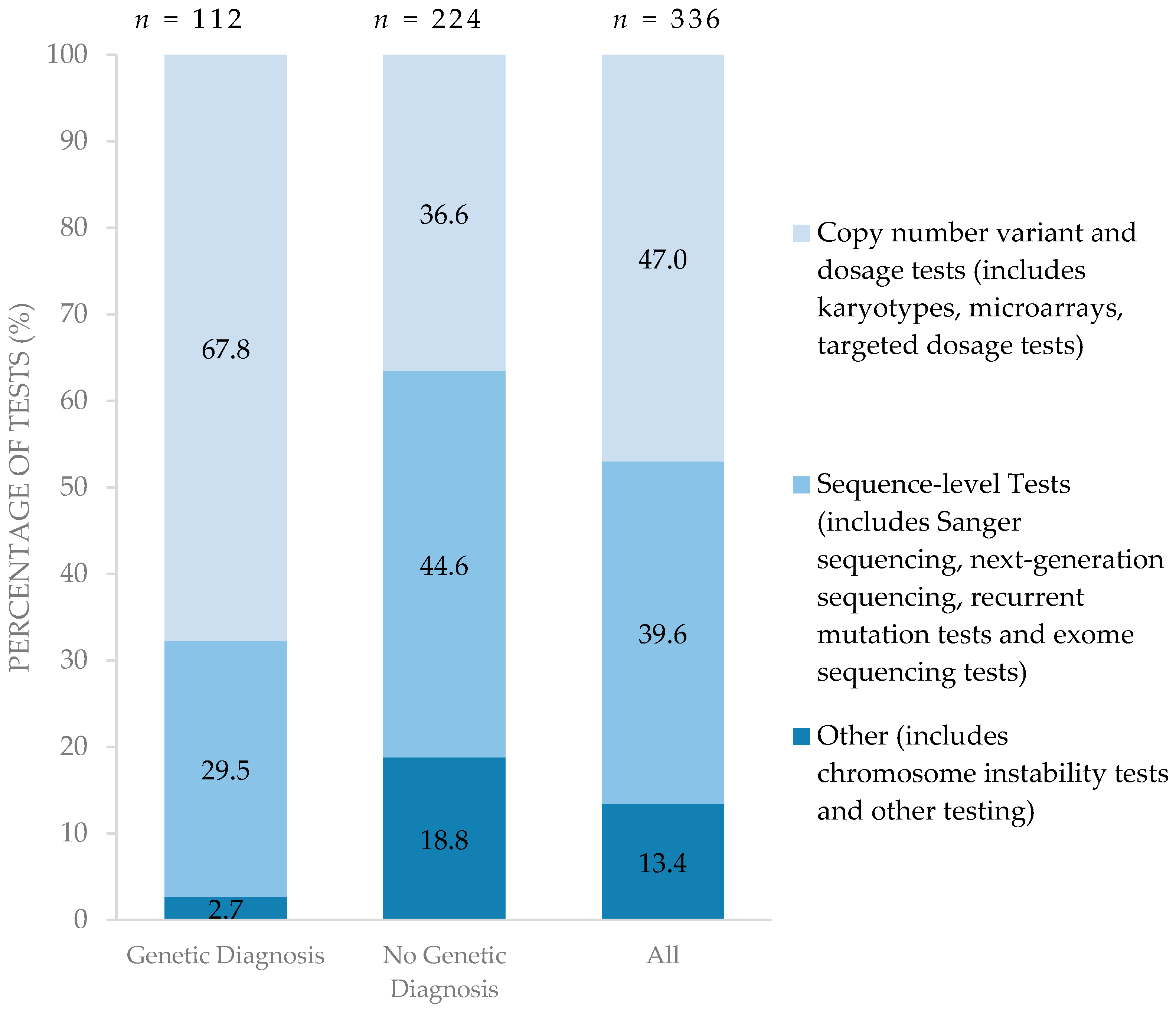{kind=link}
| Diagnosis | |||||||
|---|---|---|---|---|---|---|---|
| All Subjects | Confirmed Genetic Diagnosis | No Genetic Diagnosis | p Value 2 | ||||
| n = 63 | n = 30 | n = 33 | |||||
| Demographics 3: | 0.97 | ||||||
| Age group, n (%) | |||||||
| 0–4 years | 30 | (47) | 14 | (47) | 16 | (49) | |
| 5–9 years | 20 | (32) | 10 | (33) | 10 | (30) | |
| 10–18 years | 13 | (21) | 6 | (20) | 7 | (21) | |
| Sex, n (%) | 0.27 | ||||||
| Male | 34 | (54) | 14 | (47) | 20 | (61) | |
| Female | 29 | (46) | 16 | (53) | 13 | (39) | |
| Ethnicity, n (%) 4 | 0.03 | ||||||
| Caucasian | 17 | (27) | 5 | (17) | 12 | (36) | |
| Non-Caucasian | 30 | (48) | 19 | (63) | 11 | (33) | |
| Location | 0.59 | ||||||
| Toronto 5 | 42 | (67) | 21 | (70) | 21 | (64) | |
| Other | 21 | (33) | 9 | (30) | 12 | (36) | |
| Clinical Characteristics: | |||||||
| Prematurity 6 | 0.30 | ||||||
| Premature (<37 weeks) 1 | 12 | (19) | <5 | 8 | (24) | ||
| Term (≥37 weeks) | 50 | (79) | 25 | (83) | 25 | (76) | |
| Birth weight 7 | 0.89 | ||||||
| <2.5 kg | 15 | (24) | 7 | (23) | 8 | (24) | |
| ≥2.5 kg | 43 | (68) | 21 | (70) | 22 | (67) | |
| Developmental Delay 8 | 0.72 | ||||||
| Yes | 58 | (92) | 28 | (93) | 30 | (91) | |
| No 1 | 5 | (8) | <5 | <5 | |||
| No. organ systems involved in congenital anomalies 9 | 0.62 | ||||||
| <2 | 40 | (64) | 20 | (67) | 20 | (61) | |
| ≥2 | 23 | (36) | 10 | (33) | 13 | (39) | |
| Technology Assistance 10 | 0.69 | ||||||
| Yes | 49 | (78) | 24 | (80) | 25 | (76) | |
| No | 14 | (22) | 6 | (20) | 8 | (24) | |
| Start Date of Testing Period 11: | 0.48 | ||||||
| Prior to 1 January 2010 | 37 | (59) | 19 | (63) | 18 | (55) | |
| 1 January 2010–30 June 2014 | 26 | (41) | 11 | (37) | 15 | (45) | |
| Genetic Diagnoses in Total CCP 12 | |||||||
| Trisomy 21 | 18 | (12) | |||||
| DiGeorge syndrome | 6 | (4) | |||||
| CHARGE syndrome 13 | 5 | (4) | |||||
| Other | 118 | (80) | |||||
| All Subjects | Confirmed Genetic Diagnosis | No Genetic Diagnosis | p value 2 | ||||
|---|---|---|---|---|---|---|---|
| n = 63 | n = 30 | n = 33 | |||||
| Number of Genetic Tests | 4 | (2.5–7) | 3 | (2–4) | 6 | (4–9) | 0.002 |
| Length of Testing Period (years) | 2.31 | (0.33–6.08) | 0.35 | (0.12–3.04) | 4.12 | (1.73–8.42) | <0.001 |
| Genetic Testing Costs (C$) | 4436 | (1869–8726) | 2614 | (1605–4080) | 8496 | (4399–12,480) | <0.001 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Oei, K.; Hayeems, R.Z.; Ungar, W.J.; Cohn, R.D.; Cohen, E. Genetic Testing among Children in a Complex Care Program. Children 2017, 4, 42. https://doi.org/10.3390/children4050042
Oei K, Hayeems RZ, Ungar WJ, Cohn RD, Cohen E. Genetic Testing among Children in a Complex Care Program. Children. 2017; 4(5):42. https://doi.org/10.3390/children4050042
Chicago/Turabian StyleOei, Krista, Robin Z. Hayeems, Wendy J. Ungar, Ronald D. Cohn, and Eyal Cohen. 2017. "Genetic Testing among Children in a Complex Care Program" Children 4, no. 5: 42. https://doi.org/10.3390/children4050042