
Paediatric Thoracic Imaging in Cystic Fibrosis in the Era of Cystic Fibrosis Transmembrane Conductance Regulator Modulation
Abstract
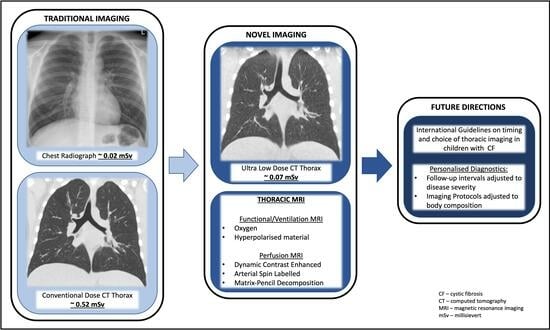
:
1. Introduction
2. Methods
3. Imaging
3.1. Chest Radiography
3.2. Computed Tomography
3.3. Magnetic Resonance Imaging
3.4. Positron Emission Tomography (PET) CT
3.5. Ultrasound
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Rowe, S.M.; Miller, S.; Sorscher, E.J. Cystic fibrosis. N. Engl. J. Med. 2005, 352, 1992–2001. [Google Scholar] [CrossRef]
- Castellani, C.; Duff, A.J.A.; Bell, S.C.; Heijerman, H.G.M.; Munck, A.; Ratjen, F.; Sermet-Gaudelus, I.; Southern, K.W.; Barben, J.; Flume, P.A.; et al. ECFS best practice guidelines: The 2018 revision. J. Cyst. Fibros. 2018, 17, 153–178. [Google Scholar] [CrossRef]
- Guo, J.; Garratt, A.; Hill, A. Worldwide rates of diagnosis and effective treatment for cystic fibrosis. J. Cyst. Fibros. 2022, 21, 456–462. [Google Scholar] [CrossRef]
- Scotet, V.; L’Hostis, C.; Ferec, C. The Changing Epidemiology of Cystic Fibrosis: Incidence, Survival and Impact of the CFTR Gene Discovery. Genes 2020, 11, 589. [Google Scholar] [CrossRef] [PubMed]
- Andersen, D.H. Cystic Fibrosis of the Pancreas and its Relation to Celiac Disease: A Clinical and Pathologic Study. Am. J. Dis. Child. 1938, 56, 344–399. [Google Scholar] [CrossRef]
- O’Connell, O.J.; McWilliams, S.; McGarrigle, A.; O’Connor, O.J.; Shanahan, F.; Mullane, D.; Eustace, J.; Maher, M.M.; Plant, B.J. Radiologic imaging in cystic fibrosis: Cumulative effective dose and changing trends over 2 decades. Chest 2012, 141, 1575–1583. [Google Scholar] [CrossRef] [PubMed]
- Silva, I.A.L.; Laselva, O.; Lopes-Pacheco, M. Advances in Preclinical In Vitro Models for the Translation of Precision Medicine for Cystic Fibrosis. J. Pers. Med. 2022, 12, 1321. [Google Scholar] [CrossRef]
- Rommens, J.M.; Iannuzzi, M.C.; Kerem, B.; Drumm, M.L.; Melmer, G.; Dean, M.; Rozmahel, R.; Cole, J.L.; Kennedy, D.; Hidaka, N.; et al. Identification of the cystic fibrosis gene: Chromosome walking and jumping. Science 1989, 245, 1059–1065. [Google Scholar] [CrossRef] [PubMed]
- Riordan, J.R.; Rommens, J.M.; Kerem, B.; Alon, N.; Rozmahel, R.; Grzelczak, Z.; Zielenski, J.; Lok, S.; Plavsic, N.; Chou, J.L.; et al. Identification of the cystic fibrosis gene: Cloning and characterization of complementary DNA. Science 1989, 245, 1066–1073. [Google Scholar] [CrossRef]
- Kerem, B.; Rommens, J.M.; Buchanan, J.A.; Markiewicz, D.; Cox, T.K.; Chakravarti, A.; Buchwald, M.; Tsui, L.C. Identification of the cystic fibrosis gene: Genetic analysis. Science 1989, 245, 1073–1080. [Google Scholar] [CrossRef]
- Saint-Criq, V.; Gray, M.A. Role of CFTR in epithelial physiology. Cell. Mol. Life Sci. 2017, 74, 93–115. [Google Scholar] [CrossRef]
- Lopes-Pacheco, M. CFTR Modulators: The Changing Face of Cystic Fibrosis in the Era of Precision Medicine. Front. Pharmacol. 2019, 10, 1662. [Google Scholar] [CrossRef]
- Lopes-Pacheco, M.; Pedemonte, N.; Veit, G. Discovery of CFTR modulators for the treatment of cystic fibrosis. Expert Opin. Drug Discov. 2021, 16, 897–913. [Google Scholar] [CrossRef]
- Lopes-Pacheco, M. CFTR Modulators: Shedding Light on Precision Medicine for Cystic Fibrosis. Front. Pharmacol. 2016, 7, 275. [Google Scholar] [CrossRef]
- Pinto, M.C.; Silva, I.A.L.; Figueira, M.F.; Amaral, M.D.; Lopes-Pacheco, M. Pharmacological Modulation of Ion Channels for the Treatment of Cystic Fibrosis. J. Exp. Pharmacol. 2021, 13, 693–723. [Google Scholar] [CrossRef]
- Mall, M.A.; Hartl, D. CFTR: Cystic fibrosis and beyond. Eur. Respir. J. 2014, 44, 1042–1054. [Google Scholar] [CrossRef] [PubMed]
- Ong, T.; Ramsey, B.W. Cystic Fibrosis: A Review. JAMA 2023, 329, 1859–1871. [Google Scholar] [CrossRef] [PubMed]
- Thia, L.P.; Calder, A.; Stocks, J.; Bush, A.; Owens, C.M.; Wallis, C.; Young, C.; Sullivan, Y.; Wade, A.; McEwan, A.; et al. Is chest CT useful in newborn screened infants with cystic fibrosis at 1 year of age? Thorax 2014, 69, 320–327. [Google Scholar] [CrossRef] [PubMed]
- Bell, S.C.; Mall, M.A.; Gutierrez, H.; Macek, M.; Madge, S.; Davies, J.C.; Burgel, P.R.; Tullis, E.; Castanos, C.; Castellani, C.; et al. The future of cystic fibrosis care: A global perspective. Lancet Respir. Med. 2020, 8, 65–124. [Google Scholar] [CrossRef]
- Wainwright, C.E.; Elborn, J.S.; Ramsey, B.W. Lumacaftor-Ivacaftor in Patients with Cystic Fibrosis Homozygous for Phe508del CFTR. N. Engl. J. Med. 2015, 373, 1783–1784. [Google Scholar] [CrossRef] [PubMed]
- Taylor-Cousar, J.L.; Munck, A.; McKone, E.F.; van der Ent, C.K.; Moeller, A.; Simard, C.; Wang, L.T.; Ingenito, E.P.; McKee, C.; Lu, Y.; et al. Tezacaftor-Ivacaftor in Patients with Cystic Fibrosis Homozygous for Phe508del. N. Engl. J. Med. 2017, 377, 2013–2023. [Google Scholar] [CrossRef]
- Heijerman, H.G.M.; McKone, E.F.; Downey, D.G.; Van Braeckel, E.; Rowe, S.M.; Tullis, E.; Mall, M.A.; Welter, J.J.; Ramsey, B.W.; McKee, C.M.; et al. Efficacy and safety of the elexacaftor plus tezacaftor plus ivacaftor combination regimen in people with cystic fibrosis homozygous for the F508del mutation: A double-blind, randomised, phase 3 trial. Lancet 2019, 394, 1940–1948. [Google Scholar] [CrossRef]
- European Medicines Agency. Ivacaftor EMA Summary of Product Characteristics. Available online: https://www.ema.europa.eu/en/documents/product-information/kalydeco-epar-product-information_en.pdf (accessed on 11 December 2023).
- Ledford, H. Cystic fibrosis drug Vertex’s latest triumph. Nat. Biotechnol. 2012, 30, 201–202. [Google Scholar] [CrossRef]
- Ramsey, B.W.; Davies, J.; McElvaney, N.G.; Tullis, E.; Bell, S.C.; Drevinek, P.; Griese, M.; McKone, E.F.; Wainwright, C.E.; Konstan, M.W.; et al. A CFTR potentiator in patients with cystic fibrosis and the G551D mutation. N. Engl. J. Med. 2011, 365, 1663–1672. [Google Scholar] [CrossRef]
- Davies, J.C.; Wainwright, C.E.; Canny, G.J.; Chilvers, M.A.; Howenstine, M.S.; Munck, A.; Mainz, J.G.; Rodriguez, S.; Li, H.; Yen, K.; et al. Efficacy and safety of ivacaftor in patients aged 6 to 11 years with cystic fibrosis with a G551D mutation. Am. J. Respir. Crit. Care Med. 2013, 187, 1219–1225. [Google Scholar] [CrossRef]
- Davies, J.C.; Cunningham, S.; Harris, W.T.; Lapey, A.; Regelmann, W.E.; Sawicki, G.S.; Southern, K.W.; Robertson, S.; Green, Y.; Cooke, J.; et al. Safety, pharmacokinetics, and pharmacodynamics of ivacaftor in patients aged 2-5 years with cystic fibrosis and a CFTR gating mutation (KIWI): An open-label, single-arm study. Lancet Respir. Med. 2016, 4, 107–115. [Google Scholar] [CrossRef] [PubMed]
- Ronan, N.J.; Einarsson, G.G.; Twomey, M.; Mooney, D.; Mullane, D.; NiChroinin, M.; O’Callaghan, G.; Shanahan, F.; Murphy, D.M.; O’Connor, O.J.; et al. CORK Study in Cystic Fibrosis: Sustained Improvements in Ultra-Low-Dose Chest CT Scores after CFTR Modulation with Ivacaftor. Chest 2018, 153, 395–403. [Google Scholar] [CrossRef] [PubMed]
- Ratjen, F.; Hug, C.; Marigowda, G.; Tian, S.; Huang, X.; Stanojevic, S.; Milla, C.E.; Robinson, P.D.; Waltz, D.; Davies, J.C.; et al. Efficacy and safety of lumacaftor and ivacaftor in patients aged 6-11 years with cystic fibrosis homozygous for F508del-CFTR: A randomised, placebo-controlled phase 3 trial. Lancet Respir. Med. 2017, 5, 557–567. [Google Scholar] [CrossRef]
- Donaldson, S.H.; Pilewski, J.M.; Griese, M.; Cooke, J.; Viswanathan, L.; Tullis, E.; Davies, J.C.; Lekstrom-Himes, J.A.; Wang, L.T.; Group, V.X.S. Tezacaftor/Ivacaftor in Subjects with Cystic Fibrosis and F508del/F508del-CFTR or F508del/G551D-CFTR. Am. J. Respir. Crit Care Med. 2018, 197, 214–224. [Google Scholar] [CrossRef]
- Food and Drug Administration Administration. Lumacaftor-ivacaftor FDA Drug Label. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2022/206038s016lbl.pdf (accessed on 11 December 2023).
- Food and Drug Administration Administration. Tezacaftor-ivacaftor FDA Drug Label. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2023/210491s011lbl.pdf (accessed on 11 December 2023).
- Middleton, P.G.; Mall, M.A.; Drevinek, P.; Lands, L.C.; McKone, E.F.; Polineni, D.; Ramsey, B.W.; Taylor-Cousar, J.L.; Tullis, E.; Vermeulen, F.; et al. Elexacaftor-Tezacaftor-Ivacaftor for Cystic Fibrosis with a Single Phe508del Allele. N. Engl. J. Med. 2019, 381, 1809–1819. [Google Scholar] [CrossRef] [PubMed]
- Bacalhau, M.; Camargo, M.; Magalhaes-Ghiotto, G.A.V.; Drumond, S.; Castelletti, C.H.M.; Lopes-Pacheco, M. Elexacaftor-Tezacaftor-Ivacaftor: A Life-Changing Triple Combination of CFTR Modulator Drugs for Cystic Fibrosis. Pharmaceuticals 2023, 16, 410. [Google Scholar] [CrossRef] [PubMed]
- Food and Drug Administration Administration. Elexacaftor-Tezacaftor-Ivacaftor FDA Drug Label. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2023/217660s000lbl.pdf (accessed on 11 December 2023).
- Medicines and Healthcare Products Regulatory Agency. Elexacaftor-Tezacaftor-Ivacaftor MHRA Summary of Product Characteristics. Available online: https://mhraproducts4853.blob.core.windows.net/docs/54f21d4e55ab2249ea23dd405af50eb8b76b5b8d (accessed on 11 December 2023).
- European Medicines Agency. Elexacaftor-Tezacaftor-Ivacaftor EMA Summary of Product Characteristics. Available online: https://www.ema.europa.eu/en/documents/product-information/kaftrio-epar-product-information_en.pdf (accessed on 11 December 2023).
- Medicines and Healthcare Products Regulatory Agency. Ivacaftor MHRA Summary of Product Characteristics. Available online: https://mhraproducts4853.blob.core.windows.net/docs/de94c1a13ce5d0f3d7dac0b0dc6102d26b2d376e (accessed on 11 December 2023).
- Food and Drug Administration Administration. Ivacaftor FDA Drug Label. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2023/203188s038lbl.pdf (accessed on 11 December 2023).
- European Medicines Agency. Lumacaftor-Ivacaftor EMA Summary of Product Characteristics. Available online: https://www.ema.europa.eu/en/documents/product-information/orkambi-epar-product-information_en.pdf (accessed on 11 December 2023).
- Medicines and Healthcare Products Regulatory Agency. Lumacaftor-Ivacaftor MHRA Summary of Product Characteristics. Available online: https://mhraproducts4853.blob.core.windows.net/docs/45ef55c039909cb939b92e95a6c58654cfd42834 (accessed on 11 December 2023).
- European Medicines Agency. Tezacaftor-Ivacaftor EMA Summary of Product Characteristics. Available online: https://www.ema.europa.eu/en/documents/product-information/symkevi-epar-product-information_en.pdf (accessed on 11 December 2023).
- Medicines and Healthcare Products Regulatory Agency. Tezacaftor-Ivacaftor MHRA Summary of Product Characteristics. Available online: https://mhraproducts4853.blob.core.windows.net/docs/c9ab16f7d588f0647f3d026352f92a6b67527417 (accessed on 11 December 2023).
- Kerem, E.; Viviani, L.; Zolin, A.; MacNeill, S.; Hatziagorou, E.; Ellemunter, H.; Drevinek, P.; Gulmans, V.; Krivec, U.; Olesen, H.; et al. Factors associated with FEV1 decline in cystic fibrosis: Analysis of the ECFS patient registry. Eur. Respir. J. 2014, 43, 125–133. [Google Scholar] [CrossRef]
- De Boeck, K. Cystic fibrosis in the year 2020: A disease with a new face. Acta Paediatr. 2020, 109, 893–899. [Google Scholar] [CrossRef]
- Greene, K.E.; Takasugi, J.E.; Godwin, J.D.; Richardson, M.L.; Burke, W.; Aitken, M.L. Radiographic changes in acute exacerbations of cystic fibrosis in adults: A pilot study. AJR Am. J. Roentgenol. 1994, 163, 557–562. [Google Scholar] [CrossRef] [PubMed]
- Proesmans, M. Best practices in the treatment of early cystic fibrosis lung disease. Ther. Adv. Respir. Dis. 2017, 11, 97–104. [Google Scholar] [CrossRef] [PubMed]
- Terheggen-Lagro, S.; Truijens, N.; van Poppel, N.; Gulmans, V.; van der Laag, J.; van der Ent, C. Correlation of six different cystic fibrosis chest radiograph scoring systems with clinical parameters. Pediatr. Pulmonol. 2003, 35, 441–445. [Google Scholar] [CrossRef] [PubMed]
- Bortoluzzi, C.F.; Pontello, E.; Pintani, E.; de Winter-de Groot, K.M.; D’Orazio, C.; Assael, B.M.; Hunink, M.G.M.; Tiddens, H.; Caudri, D.; Group, C.F.C.S. The impact of chest computed tomography and chest radiography on clinical management of cystic fibrosis lung disease. J. Cyst. Fibros. 2020, 19, 641–646. [Google Scholar] [CrossRef]
- Zucker, E.J.; Barnes, Z.A.; Lungren, M.P.; Shpanskaya, Y.; Seekins, J.M.; Halabi, S.S.; Larson, D.B. Deep learning to automate Brasfield chest radiographic scoring for cystic fibrosis. J. Cyst. Fibros. 2020, 19, 131–138. [Google Scholar] [CrossRef]
- Goralski, J.L.; Stewart, N.J.; Woods, J.C. Novel imaging techniques for cystic fibrosis lung disease. Pediatr. Pulmonol. 2021, 56 (Suppl. S1), S40–S54. [Google Scholar] [CrossRef]
- Hino, T.; Hata, A.; Hida, T.; Yamada, Y.; Ueyama, M.; Araki, T.; Kamitani, T.; Nishino, M.; Kurosaki, A.; Jinzaki, M.; et al. Projected lung areas using dynamic X-ray (DXR). Eur. J. Radiol. Open 2020, 7, 100263. [Google Scholar] [CrossRef]
- Hida, T.; Yamada, Y.; Ueyama, M.; Araki, T.; Nishino, M.; Kurosaki, A.; Jinzaki, M.; Honda, H.; Hatabu, H.; Kudoh, S. Time-resolved quantitative evaluation of diaphragmatic motion during forced breathing in a health screening cohort in a standing position: Dynamic chest phrenicography. Eur. J. Radiol. 2019, 113, 59–65. [Google Scholar] [CrossRef]
- FitzMaurice, T.S.; McCann, C.; Nazareth, D.; Shaw, M.; McNamara, P.S.; Walshaw, M.J. Measuring the effect of elexacaftor/tezacaftor/ivacaftor combination therapy on the respiratory pump in people with CF using dynamic chest radiography. J. Cyst. Fibros. 2022, 21, 1036–1041. [Google Scholar] [CrossRef]
- Martínez, T.M.; Llapur, C.J.; Williams, T.H.; Coates, C.; Gunderman, R.; Cohen, M.D.; Howenstine, M.S.; Saba, O.; Coxson, H.O.; Tepper, R.S. High-resolution computed tomography imaging of airway disease in infants with cystic fibrosis. Am. J. Respir. Crit. Care Med. 2005, 172, 1133–1138. [Google Scholar] [CrossRef]
- Mott, L.S.; Park, J.; Murray, C.P.; Gangell, C.L.; de Klerk, N.H.; Robinson, P.J.; Robertson, C.F.; Ranganathan, S.C.; Sly, P.D.; Stick, S.M. Progression of early structural lung disease in young children with cystic fibrosis assessed using CT. Thorax 2012, 67, 509–516. [Google Scholar] [CrossRef]
- de Jong, P.A.; Nakano, Y.; Lequin, M.H.; Mayo, J.R.; Woods, R.; Paré, P.D.; Tiddens, H.A. Progressive damage on high resolution computed tomography despite stable lung function in cystic fibrosis. Eur. Respir. J. 2004, 23, 93–97. [Google Scholar] [CrossRef]
- Pillarisetti, N.; Williamson, E.; Linnane, B.; Skoric, B.; Robertson, C.F.; Robinson, P.; Massie, J.; Hall, G.L.; Sly, P.; Stick, S.; et al. Infection, inflammation, and lung function decline in infants with cystic fibrosis. Am. J. Respir. Crit. Care Med. 2011, 184, 75–81. [Google Scholar] [CrossRef]
- Gustafsson, P.M.; De Jong, P.A.; Tiddens, H.A.; Lindblad, A. Multiple-breath inert gas washout and spirometry versus structural lung disease in cystic fibrosis. Thorax 2008, 63, 129–134. [Google Scholar] [CrossRef]
- Stick, S.M.; Brennan, S.; Murray, C.; Douglas, T.; von Ungern-Sternberg, B.S.; Garratt, L.W.; Gangell, C.L.; De Klerk, N.; Linnane, B.; Ranganathan, S.; et al. Bronchiectasis in infants and preschool children diagnosed with cystic fibrosis after newborn screening. J. Pediatr. 2009, 155, 623–628.e1. [Google Scholar] [CrossRef]
- Wijker, N.E.; Vidmar, S.; Grimwood, K.; Sly, P.D.; Byrnes, C.A.; Carlin, J.B.; Cooper, P.J.; Robertson, C.F.; Massie, R.J.; Kemner van de Corput, M.P.C.; et al. Early markers of cystic fibrosis structural lung disease: Follow-up of the ACFBAL cohort. Eur. Respir. J. 2020, 55, 1901694. [Google Scholar] [CrossRef]
- Nissenbaum, C.; Davies, G.; Horsley, A.; Davies, J.C. Monitoring early stage lung disease in cystic fibrosis. Curr. Opin. Pulm. Med. 2020, 26, 671–678. [Google Scholar] [CrossRef]
- Cademartiri, F.; Luccichenti, G.; Palumbo, A.A.; Maffei, E.; Pisi, G.; Zompatori, M.; Krestin, G.P. Predictive value of chest CT in patients with cystic fibrosis: A single-center 10-year experience. AJR Am. J. Roentgenol. 2008, 190, 1475–1480. [Google Scholar] [CrossRef]
- Brody, A.S.; Klein, J.S.; Molina, P.L.; Quan, J.; Bean, J.A.; Wilmott, R.W. High-resolution computed tomography in young patients with cystic fibrosis: Distribution of abnormalities and correlation with pulmonary function tests. J. Pediatr. 2004, 145, 32–38. [Google Scholar] [CrossRef]
- Sanders, D.B.; Li, Z.; Brody, A.S.; Farrell, P.M. Chest computed tomography scores of severity are associated with future lung disease progression in children with cystic fibrosis. Am. J. Respir. Crit. Care Med. 2011, 184, 816–821. [Google Scholar] [CrossRef]
- Loeve, M.; Hop, W.C.; de Bruijne, M.; van Hal, P.T.; Robinson, P.; Aitken, M.L.; Dodd, J.D.; Tiddens, H.A. Chest computed tomography scores are predictive of survival in patients with cystic fibrosis awaiting lung transplantation. Am. J. Respir. Crit. Care Med. 2012, 185, 1096–1103. [Google Scholar] [CrossRef]
- Bhalla, M.; Turcios, N.; Aponte, V.; Jenkins, M.; Leitman, B.S.; McCauley, D.I.; Naidich, D.P. Cystic fibrosis: Scoring system with thin-section CT. Radiology 1991, 179, 783–788. [Google Scholar] [CrossRef]
- Rosenow, T.; Oudraad, M.C.; Murray, C.P.; Turkovic, L.; Kuo, W.; de Bruijne, M.; Ranganathan, S.C.; Tiddens, H.A.; Stick, S.M. PRAGMA-CF. A Quantitative Structural Lung Disease Computed Tomography Outcome in Young Children with Cystic Fibrosis. Am. J. Respir. Crit. Care Med. 2015, 191, 1158–1165. [Google Scholar] [CrossRef] [PubMed]
- Mok, L.C.; Garcia-Uceda, A.; Cooper, M.N.; Kemner-Van De Corput, M.; De Bruijne, M.; Feyaerts, N.; Rosenow, T.; De Boeck, K.; Stick, S.; Tiddens, H. The effect of CFTR modulators on structural lung disease in cystic fibrosis. Front. Pharmacol. 2023, 14, 1147348. [Google Scholar] [CrossRef] [PubMed]
- Dournes, G.; Hall, C.S.; Willmering, M.M.; Brody, A.S.; Macey, J.; Bui, S.; Denis de Senneville, B.; Berger, P.; Laurent, F.; Benlala, I.; et al. Artificial intelligence in computed tomography for quantifying lung changes in the era of CFTR modulators. Eur. Respir. J. 2022, 59, 2100844. [Google Scholar] [CrossRef]
- Lundstrom, C.F.; Gilmore, H.L.; Ros, P.R. Integrated Diagnostics: The Computational Revolution Catalyzing Cross-disciplinary Practices in Radiology, Pathology, and Genomics. Radiology 2017, 285, 12–15. [Google Scholar] [CrossRef]
- Perez-Rovira, A.; Kuo, W.; Petersen, J.; Tiddens, H.A.; de Bruijne, M. Automatic airway-artery analysis on lung CT to quantify airway wall thickening and bronchiectasis. Med. Phys. 2016, 43, 5736. [Google Scholar] [CrossRef]
- Schalekamp, S.; Klein, W.M.; van Leeuwen, K.G. Current and emerging artificial intelligence applications in chest imaging: A pediatric perspective. Pediatr. Radiol. 2022, 52, 2120–2130. [Google Scholar] [CrossRef]
- Crowley, C.; Connor, O.J.O.; Ciet, P.; Tiddens, H.; Maher, M.M. The evolving role of radiological imaging in cystic fibrosis. Curr. Opin. Pulm. Med. 2021, 27, 575–585. [Google Scholar] [CrossRef] [PubMed]
- Ferris, H.; Twomey, M.; Moloney, F.; O’Neill, S.B.; Murphy, K.; O’Connor, O.J.; Maher, M. Computed tomography dose optimisation in cystic fibrosis: A review. World J. Radiol. 2016, 8, 331–341. [Google Scholar] [CrossRef] [PubMed]
- Katsura, M.; Matsuda, I.; Akahane, M.; Sato, J.; Akai, H.; Yasaka, K.; Kunimatsu, A.; Ohtomo, K. Model-based iterative reconstruction technique for radiation dose reduction in chest CT: Comparison with the adaptive statistical iterative reconstruction technique. Eur. Radiol. 2012, 22, 1613–1623. [Google Scholar] [CrossRef] [PubMed]
- Miéville, F.A.; Berteloot, L.; Grandjean, A.; Ayestaran, P.; Gudinchet, F.; Schmidt, S.; Brunelle, F.; Bochud, F.O.; Verdun, F.R. Model-based iterative reconstruction in pediatric chest CT: Assessment of image quality in a prospective study of children with cystic fibrosis. Pediatr. Radiol. 2013, 43, 558–567. [Google Scholar] [CrossRef] [PubMed]
- Neroladaki, A.; Botsikas, D.; Boudabbous, S.; Becker, C.D.; Montet, X. Computed tomography of the chest with model-based iterative reconstruction using a radiation exposure similar to chest X-ray examination: Preliminary observations. Eur. Radiol. 2013, 23, 360–366. [Google Scholar] [CrossRef] [PubMed]
- Loeve, M.; Lequin, M.H.; de Bruijne, M.; Hartmann, I.J.; Gerbrands, K.; van Straten, M.; Hop, W.C.; Tiddens, H.A. Cystic fibrosis: Are volumetric ultra-low-dose expiratory CT scans sufficient for monitoring related lung disease? Radiology 2009, 253, 223–229. [Google Scholar] [CrossRef] [PubMed]
- Arakawa, H.; Webb, W.R. Air trapping on expiratory high-resolution CT scans in the absence of inspiratory scan abnormalities: Correlation with pulmonary function tests and differential diagnosis. AJR Am. J. Roentgenol. 1998, 170, 1349–1353. [Google Scholar] [CrossRef] [PubMed]
- Tiddens, H.A. Chest computed tomography scans should be considered as a routine investigation in cystic fibrosis. Paediatr. Respir. Rev. 2006, 7, 202–208. [Google Scholar] [CrossRef]
- Wu, Y.; Ye, Z.; Chen, J.; Deng, L.; Song, B. Photon Counting CT: Technical Principles, Clinical Applications, and Future Prospects. Acad. Radiol. 2023, 30, 2362–2382. [Google Scholar] [CrossRef]
- Willemink, M.J.; Persson, M.; Pourmorteza, A.; Pelc, N.J.; Fleischmann, D. Photon-counting CT: Technical Principles and Clinical Prospects. Radiology 2018, 289, 293–312. [Google Scholar] [CrossRef] [PubMed]
- Donadieu, J.; Roudier, C.; Saguintaah, M.; Maccia, C.; Chiron, R. Estimation of the radiation dose from thoracic CT scans in a cystic fibrosis population. Chest 2007, 132, 1233–1238. [Google Scholar] [CrossRef]
- Joyce, S.; Carey, B.W.; Moore, N.; Mullane, D.; Moore, M.; McEntee, M.F.; Plant, B.J.; Maher, M.M.; O’Connor, O.J. Computed tomography in cystic fibrosis lung disease: A focus on radiation exposure. Pediatr. Radiol. 2021, 51, 544–553. [Google Scholar] [CrossRef]
- van Straten, M.; Brody, A.S.; Ernst, C.; Guillerman, R.P.; Tiddens, H.; Nagle, S.K. Guidance for computed tomography (CT) imaging of the lungs for patients with cystic fibrosis (CF) in research studies. J. Cyst. Fibros. 2020, 19, 176–183. [Google Scholar] [CrossRef]
- Sheahan, K.P.; O’Mahony, A.T.; Morrissy, D.; Ibrahim, H.; Crowley, C.; Waldron, M.G.; Sokol-Randell, D.; McMahon, A.; Maher, M.M.; O’Connor, O.J.; et al. Replacing Plain Radiograph with ultra-low dose CT thorax in cystic fibrosis (CF) in the era of CFTR modulation and its impact on cumulative effective dose. J. Cyst. Fibros. 2023, 22, 715–721. [Google Scholar] [CrossRef]
- Delacoste, J.; Chaptinel, J.; Beigelman-Aubry, C.; Piccini, D.; Sauty, A.; Stuber, M. A double echo ultra short echo time (UTE) acquisition for respiratory motion-suppressed high resolution imaging of the lung. Magn. Reson. Med. 2018, 79, 2297–2305. [Google Scholar] [CrossRef] [PubMed]
- Dournes, G.; Menut, F.; Macey, J.; Fayon, M.; Chateil, J.F.; Salel, M.; Corneloup, O.; Montaudon, M.; Berger, P.; Laurent, F. Lung morphology assessment of cystic fibrosis using MRI with ultra-short echo time at submillimeter spatial resolution. Eur. Radiol. 2016, 26, 3811–3820. [Google Scholar] [CrossRef] [PubMed]
- Roach, D.J.; Crémillieux, Y.; Fleck, R.J.; Brody, A.S.; Serai, S.D.; Szczesniak, R.D.; Kerlakian, S.; Clancy, J.P.; Woods, J.C. Ultrashort Echo-Time Magnetic Resonance Imaging Is a Sensitive Method for the Evaluation of Early Cystic Fibrosis Lung Disease. Ann. Am. Thorac. Soc. 2016, 13, 1923–1931. [Google Scholar] [CrossRef] [PubMed]
- Wielpütz, M.O.; von Stackelberg, O.; Stahl, M.; Jobst, B.J.; Eichinger, M.; Puderbach, M.U.; Nährlich, L.; Barth, S.; Schneider, C.; Kopp, M.V.; et al. Multicentre standardisation of chest MRI as radiation-free outcome measure of lung disease in young children with cystic fibrosis. J. Cyst. Fibros. 2018, 17, 518–527. [Google Scholar] [CrossRef]
- Stahl, M.; Steinke, E.; Graeber, S.Y.; Joachim, C.; Seitz, C.; Kauczor, H.U.; Eichinger, M.; Hämmerling, S.; Sommerburg, O.; Wielpütz, M.O.; et al. Magnetic Resonance Imaging Detects Progression of Lung Disease and Impact of Newborn Screening in Preschool Children with Cystic Fibrosis. Am. J. Respir. Crit. Care Med. 2021, 204, 943–953. [Google Scholar] [CrossRef]
- Shammi, U.A.; D’Alessandro, M.F.; Altes, T.; Hersman, F.W.; Ruset, I.C.; Mugler, J., 3rd; Meyer, C.; Mata, J.; Qing, K.; Thomen, R. Comparison of Hyperpolarized (3)He and (129)Xe MR Imaging in Cystic Fibrosis Patients. Acad. Radiol. 2022, 29 (Suppl. S2), S82–S90. [Google Scholar] [CrossRef]
- Kern, A.L.; Vogel-Claussen, J. Hyperpolarized gas MRI in pulmonology. Br. J. Radiol. 2018, 91, 20170647. [Google Scholar] [CrossRef]
- Wang, Z.J.; Ohliger, M.A.; Larson, P.E.Z.; Gordon, J.W.; Bok, R.A.; Slater, J.; Villanueva-Meyer, J.E.; Hess, C.P.; Kurhanewicz, J.; Vigneron, D.B. Hyperpolarized (13)C MRI: State of the Art and Future Directions. Radiology 2019, 291, 273–284. [Google Scholar] [CrossRef]
- Jakob, P.M.; Wang, T.; Schultz, G.; Hebestreit, H.; Hebestreit, A.; Hahn, D. Assessment of human pulmonary function using oxygen-enhanced T(1) imaging in patients with cystic fibrosis. Magn. Reson. Med. 2004, 51, 1009–1016. [Google Scholar] [CrossRef]
- Ohno, Y.; Hatabu, H. Basics concepts and clinical applications of oxygen-enhanced MR imaging. Eur. J. Radiol. 2007, 64, 320–328. [Google Scholar] [CrossRef]
- Neemuchwala, F.; Ghadimi Mahani, M.; Pang, Y.; Lee, E.; Johnson, T.D.; Galbán, C.J.; Fortuna, A.B.; Sanchez-Jacob, R.; Flask, C.A.; Nasr, S.Z. Lung T1 mapping magnetic resonance imaging in the assessment of pulmonary disease in children with cystic fibrosis: A pilot study. Pediatr. Radiol. 2020, 50, 923–934. [Google Scholar] [CrossRef]
- McBennett, K.; MacAskill, C.J.; Keshock, E.; Mahani, M.G.; Mata, J.; Towbin, A.J.; Sankararaman, S.; Drumm, M.L.; Yu, X.; Ren, C.L.; et al. Magnetic resonance imaging of cystic fibrosis: Multi-organ imaging in the age of CFTR modulator therapies. J. Cyst. Fibros. 2022, 21, e148–e157. [Google Scholar] [CrossRef] [PubMed]
- Heidenreich, J.F.; Kuhl, P.J.; Grunz, J.P.; Hendel, R.; Metz, C.; Weng, A.M.; Benkert, T.; Hebestreit, H.; Bley, T.A.; Kostler, H.; et al. Lung Function in Patients with Cystic Fibrosis before and during CFTR-Modulator Therapy Using 3D Ultrashort Echo Time MRI. Radiology 2023, 308, e230084. [Google Scholar] [CrossRef] [PubMed]
- Wagener, J.S.; Williams, M.J.; Millar, S.J.; Morgan, W.J.; Pasta, D.J.; Konstan, M.W. Pulmonary exacerbations and acute declines in lung function in patients with cystic fibrosis. J. Cyst. Fibros. 2018, 17, 496–502. [Google Scholar] [CrossRef] [PubMed]
- Streibel, C.; Willers, C.C.; Pusterla, O.; Bauman, G.; Stranzinger, E.; Brabandt, B.; Bieri, O.; Curdy, M.; Bullo, M.; Frauchiger, B.S.; et al. Effects of elexacaftor/tezacaftor/ivacaftor therapy in children with cystic fibrosis—A comprehensive assessment using lung clearance index, spirometry, and functional and structural lung MRI. J. Cyst. Fibros. 2023, 22, 615–622. [Google Scholar] [CrossRef]
- Mai, V.M.; Berr, S.S. MR perfusion imaging of pulmonary parenchyma using pulsed arterial spin labeling techniques: FAIRER and FAIR. J. Magn. Reson. Imaging 1999, 9, 483–487. [Google Scholar] [CrossRef]
- Graeber, S.Y.; Renz, D.M.; Stahl, M.; Pallenberg, S.T.; Sommerburg, O.; Naehrlich, L.; Berges, J.; Dohna, M.; Ringshausen, F.C.; Doellinger, F.; et al. Effects of Elexacaftor/Tezacaftor/Ivacaftor Therapy on Lung Clearance Index and Magnetic Resonance Imaging in Patients with Cystic Fibrosis and One or Two F508del Alleles. Am. J. Respir. Crit. Care Med. 2022, 206, 311–320. [Google Scholar] [CrossRef] [PubMed]
- Fainardi, V.; Skenderaj, K.; Ciuni, A.; Milanese, G.; Deolmi, M.; Longo, F.; Spaggiari, C.; Sverzellati, N.; Esposito, S.; Pisi, G. Structural changes in lung morphology detected by MRI after modulating therapy with elexacaftor/tezacaftor/ivacaftor in adolescent and adult patients with cystic fibrosis. Respir. Med. 2023, 216, 107328. [Google Scholar] [CrossRef] [PubMed]
- Masand, P.M.; Narkewicz, M.R.; Leung, D.H. The Emergence of Elastography for Cystic Fibrosis Liver Disease. J. Cyst. Fibros. 2020, 19, 339–341. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Liu, H.; Li, X.; Zhou, L.; Wang, R.; Zhang, Y. Application of BOLD-MRI in the classification of renal function in chronic kidney disease. Abdom. Radiol. 2019, 44, 604–611. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, J.A.; Kramer, C.M. Role of Imaging Techniques for Diagnosis, Prognosis and Management of Heart Failure Patients: Cardiac Magnetic Resonance. Curr. Heart Fail. Rep. 2015, 12, 276–283. [Google Scholar] [CrossRef]
- Sequeiros, I.M.; Hester, K.; Callaway, M.; Williams, A.; Garland, Z.; Powell, T.; Wong, F.S.; Jarad, N.A. MRI appearance of the pancreas in patients with cystic fibrosis: A comparison of pancreas volume in diabetic and non-diabetic patients. Br. J. Radiol. 2010, 83, 921–926. [Google Scholar] [CrossRef]
- Klein, M.; Cohen-Cymberknoh, M.; Armoni, S.; Shoseyov, D.; Chisin, R.; Orevi, M.; Freedman, N.; Kerem, E. 18F-fluorodeoxyglucose-PET/CT imaging of lungs in patients with cystic fibrosis. Chest 2009, 136, 1220–1228. [Google Scholar] [CrossRef]
- Amin, R.; Charron, M.; Grinblat, L.; Shammas, A.; Grasemann, H.; Graniel, K.; Ciet, P.; Tiddens, H.; Ratjen, F. Cystic fibrosis: Detecting changes in airway inflammation with FDG PET/CT. Radiology 2012, 264, 868–875. [Google Scholar] [CrossRef]
- Zhang, Q.; Hu, Y.; Zhou, C.; Zhao, Y.; Zhang, N.; Zhou, Y.; Yang, Y.; Zheng, H.; Fan, W.; Liang, D.; et al. Reducing pediatric total-body PET/CT imaging scan time with multimodal artificial intelligence technology. EJNMMI Phys. 2024, 11, 1. [Google Scholar] [CrossRef]
- Sansone, F.; Attanasi, M.; Di Filippo, P.; Sferrazza Papa, G.F.; Di Pillo, S.; Chiarelli, F. Usefulness of Lung Ultrasound in Paediatric Respiratory Diseases. Diagnostics 2021, 11, 1783. [Google Scholar] [CrossRef] [PubMed]
- Curatola, A.; Corona, F.; Squillaci, D.; Saccari, A.; Chiaretti, A.; Barbi, E.; Maschio, M. Lung ultrasound evaluation in people with cystic fibrosis: A new approach in the pulmonology outpatient clinic. Pediatr. Pulmonol. 2023, 59, 592–599. [Google Scholar] [CrossRef] [PubMed]
- Volpicelli, G.; Elbarbary, M.; Blaivas, M.; Lichtenstein, D.A.; Mathis, G.; Kirkpatrick, A.W.; Melniker, L.; Gargani, L.; Noble, V.E.; Via, G.; et al. International evidence-based recommendations for point-of-care lung ultrasound. Intensive Care Med. 2012, 38, 577–591. [Google Scholar] [CrossRef] [PubMed]
- Peixoto, A.O.; Marson, F.A.; Dertkigil, S.S.; Dertkigil, R.P.; Souza, T.H.; Fraga, A.M.; Ribeiro, A.F.; Toro, A.A.; Ribeiro, J.D. The Use of Ultrasound as a Tool to Evaluate Pulmonary Disease in Cystic Fibrosis. Respir. Care 2020, 65, 293–303. [Google Scholar] [CrossRef]
- Strzelczuk-Judka, L.; Wojsyk-Banaszak, I.; Zakrzewska, A.; Jonczyk-Potoczna, K. Diagnostic value of chest ultrasound in children with cystic fibrosis—Pilot study. PLoS ONE 2019, 14, e0215786. [Google Scholar] [CrossRef]
- Kazmerski, T.M.; Gmelin, T.; Slocum, B.; Borrero, S.; Miller, E. Attitudes and Decision Making Related to Pregnancy among Young Women with Cystic Fibrosis. Matern. Child Health J. 2017, 21, 818–824. [Google Scholar] [CrossRef] [PubMed]
- O’Connor, K.E.; Goodwin, D.L.; NeSmith, A.; Garcia, B.; Mingora, C.; Ladores, S.L.; Rowe, S.M.; Krick, S.; Solomon, G.M. Elexacafator/tezacaftor/ivacaftor resolves subfertility in females with CF: A two center case series. J. Cyst. Fibros. 2021, 20, 399–401. [Google Scholar] [CrossRef]
- Collins, B.; Fortner, C.; Cotey, A.; Esther, C.R.J.; Trimble, A. Drug exposure to infants born to mothers taking Elexacaftor, Tezacaftor, and Ivacaftor. J. Cyst. Fibros. 2022, 21, 725–727. [Google Scholar] [CrossRef]
- Wood, B.P. Cystic fibrosis: 1997. Radiology 1997, 204, 1–10. [Google Scholar] [CrossRef]
- Weiser, G.; Kerem, E. Early intervention in CF: How to monitor the effect. Pediatr. Pulmonol. 2007, 42, 1002–1007. [Google Scholar] [CrossRef]
- Despotes, K.A.; Donaldson, S.H. Current state of CFTR modulators for treatment of Cystic Fibrosis. Curr. Opin. Pharmacol. 2022, 65, 102239. [Google Scholar] [CrossRef] [PubMed]
- Stranneheim, H.; Wedell, A. Exome and genome sequencing: A revolution for the discovery and diagnosis of monogenic disorders. J. Intern. Med. 2016, 279, 3–15. [Google Scholar] [CrossRef] [PubMed]
- Winkler, E.C.; Knoppers, B.M. Ethical challenges of precision cancer medicine. Semin. Cancer Biol. 2022, 84, 263–270. [Google Scholar] [CrossRef] [PubMed]
- Normanno, N.; Apostolidis, K.; Wolf, A.; Al Dieri, R.; Deans, Z.; Fairley, J.; Maas, J.; Martinez, A.; Moch, H.; Nielsen, S.; et al. Access and quality of biomarker testing for precision oncology in Europe. Eur. J. Cancer 2022, 176, 70–77. [Google Scholar] [CrossRef]
- Stenzinger, A.; Edsjö, A.; Ploeger, C.; Friedman, M.; Fröhling, S.; Wirta, V.; Seufferlein, T.; Botling, J.; Duyster, J.; Akhras, M.; et al. Trailblazing precision medicine in Europe: A joint view by Genomic Medicine Sweden and the Centers for Personalized Medicine, ZPM, in Germany. Semin. Cancer Biol. 2022, 84, 242–254. [Google Scholar] [CrossRef]
- Ibrahim, H.; Danish, H.; Morrissey, D.; Deasy, K.F.; McCarthy, M.; Dorgan, J.; Fleming, C.; Howlett, C.; Twohig, S.; Vagg, T.; et al. Individualized approach to elexacaftor/tezacaftor/ivacaftor dosing in cystic fibrosis, in response to self-reported anxiety and neurocognitive adverse events: A case series. Front. Pharmacol. 2023, 14, 1156621. [Google Scholar] [CrossRef]
- Hong, E.; Li, R.; Shi, A.; Almond, L.M.; Wang, J.; Khudari, A.Z.; Haddad, S.; Sislyan, S.; Angelich, M.; Chung, P.S.; et al. Safety of elexacaftor/tezacaftor/ivacaftor dose reduction: Mechanistic exploration through physiologically based pharmacokinetic modeling and a clinical case series. Pharmacotherapy 2023, 43, 291–299. [Google Scholar] [CrossRef]
- Spoletini, G.; Gillgrass, L.; Pollard, K.; Shaw, N.; Williams, E.; Etherington, C.; Clifton, I.J.; Peckham, D.G. Dose adjustments of Elexacaftor/Tezacaftor/Ivacaftor in response to mental health side effects in adults with cystic fibrosis. J. Cyst. Fibros. 2022, 21, 1061–1065. [Google Scholar] [CrossRef]
- Tiddens, H.A.; Stick, S.M.; Wild, J.M.; Ciet, P.; Parker, G.J.; Koch, A.; Vogel-Claussen, J. Respiratory tract exacerbations revisited: Ventilation, inflammation, perfusion, and structure (VIPS) monitoring to redefine treatment. Pediatr. Pulmonol. 2015, 50 (Suppl. S40), S57–S65. [Google Scholar] [CrossRef]
- Sanders, D.B.; Bittner, R.C.; Rosenfeld, M.; Redding, G.J.; Goss, C.H. Pulmonary exacerbations are associated with subsequent FEV1 decline in both adults and children with cystic fibrosis. Pediatr. Pulmonol. 2011, 46, 393–400. [Google Scholar] [CrossRef]
- Moloney, F.; Kavanagh, R.G.; Ronan, N.J.; Grey, T.M.; Joyce, S.; Ryan, D.J.; Moore, N.; O’Connor, O.J.; Plant, B.J.; Maher, M.M. Ultra-low-dose thoracic CT with model-based iterative reconstruction (MBIR) in cystic fibrosis patients undergoing treatment with cystic fibrosis transmembrane conductance regulators (CFTR). Clin. Radiol. 2021, 76, 393.e9–393.e17. [Google Scholar] [CrossRef] [PubMed]
- Maisonneuve, P.; Marshall, B.C.; Knapp, E.A.; Lowenfels, A.B. Cancer risk in cystic fibrosis: A 20-year nationwide study from the United States. J. Natl. Cancer Inst. 2013, 105, 122–129. [Google Scholar] [CrossRef] [PubMed]
- Dorneles, C.M.; Pacini, G.S.; Zanon, M.; Altmayer, S.; Watte, G.; Barros, M.C.; Marchiori, E.; Baldisserotto, M.; Hochhegger, B. Ultra-low-dose chest computed tomography without anesthesia in the assessment of pediatric pulmonary diseases. J. Pediatr. 2020, 96, 92–99. [Google Scholar] [CrossRef] [PubMed]
- Brink, J.A. Dose tracking and rational examination selection for the medically-exposed population. Health Phys. 2014, 106, 225–228. [Google Scholar] [CrossRef] [PubMed]
- AlSuwaidi, J.S.; AlBalooshi, L.G.; AlAwadhi, H.M.; Rahanjam, A.; ElHallag, M.A.; Ibrahim, J.S.; Rehani, M.M. Continuous monitoring of CT dose indexes at Dubai Hospital. AJR Am. J. Roentgenol. 2013, 201, 858–864. [Google Scholar] [CrossRef]
- O’Neill, S.; Glynn, D.; Murphy, K.P.; James, K.; Twomey, M.; Kavanagh, R.; O’Connor, O.J.; Maher, M.M. An Assessment of the Quality of CT Radiation Dose Information on the Internet. J. Am. Coll. Radiol. 2018, 15, 11–18. [Google Scholar] [CrossRef]
- Conway, S.; Balfour-Lynn, I.M.; De Rijcke, K.; Drevinek, P.; Foweraker, J.; Havermans, T.; Heijerman, H.; Lannefors, L.; Lindblad, A.; Macek, M.; et al. European Cystic Fibrosis Society Standards of Care: Framework for the Cystic Fibrosis Centre. J. Cyst. Fibros. 2014, 13 (Suppl. S1), S3–S22. [Google Scholar] [CrossRef]
- Southern, K.; Addy, C.; Bell, S.; Bevan, A.; Borawska, U.; Brown, C.; Burgel, P.R.; Button, B.; Castellani, C.; Chansard, A.; et al. Standards for the care of people with cystic fibrosis; establishing and maintaining health. J. Cyst. Fibros. 2023; in press. [Google Scholar] [CrossRef]
- Rowbotham, N.J.; Smith, S.; Leighton, P.A.; Rayner, O.C.; Gathercole, K.; Elliott, Z.C.; Nash, E.F.; Daniels, T.; Duff, A.J.A.; Collins, S.; et al. The top 10 research priorities in cystic fibrosis developed by a partnership between people with CF and healthcare providers. Thorax 2018, 73, 388–390. [Google Scholar] [CrossRef]
- Bayfield, K.J.; Douglas, T.A.; Rosenow, T.; Davies, J.C.; Elborn, S.J.; Mall, M.; Paproki, A.; Ratjen, F.; Sly, P.D.; Smyth, A.R.; et al. Time to get serious about the detection and monitoring of early lung disease in cystic fibrosis. Thorax 2021, 76, 1255–1265. [Google Scholar] [CrossRef]
- Ciet, P.; Bertolo, S.; Ros, M.; Casciaro, R.; Cipolli, M.; Colagrande, S.; Costa, S.; Galici, V.; Gramegna, A.; Lanza, C.; et al. State-of-the-art review of lung imaging in cystic fibrosis with recommendations for pulmonologists and radiologists from the “iMAging managEment of cySTic fibROsis” (MAESTRO) consortium. Eur. Respir. Rev. 2022, 31, 210173. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| CFTR Modulator | Mutations Targeted | EMA | FDA | MHRA |
|---|---|---|---|---|
| Ivacaftor (KALYDECO) (improves the activity of the defective CFTR protein) | Indicated for use in CF patients with R117H CFTR mutation or one of the following gating (class III) mutations in the CFTR gene: G551D, G1244E, G1349D, G178R, G551S, S1251N, S1255P, S549N or S549R [23,38,39]. | First approval—2012 Currently approved for infants at least 4 months and > 5 kg [23]. | First approval—2012 Currently approved for infants at least 1 month [39]. | First approval—2012 Currently approved for infants at least one month, toddlers and children >3 kg [38]. |
| Lumacaftor–ivacaftor (ORKAMBI) (lumacaftor increases the number of CFTR proteins on the cell surface) | Indicated in patients who are homozygous for the F508del mutation in the CFTR gene [31,40,41]. | First approval—2015 Currently approved for patients 1 year and older [40]. | First approval—2015 Currently approved for patients 1 year and older [31]. | First approval—2015 Currently approved for patients 1 year and older [41]. |
| Tezacaftor–ivacaftor (SYMKEVI) (tezacaftor increases the number of CFTR proteins on the cell surface) | Indicated in patients homozygous for the F508del mutation or who are heterozygous for the F508del mutation and have one of the following mutations in the (CFTR) gene: P67L, R117C, L206W, R352Q, A455E, D579G, 711 + 3A → G, S945L, S977F, R1070W, D1152H, 2789 + 5G → A, 3272-26A → G and 3849 + 10kbC → T [32,42,43]. | First approval—2018 Currently approved for patients 6 years and older [42]. | First approval—2018 Currently approved for patients 6 years and older [32]. | First approval—2018 Currently approved for patients 6 years and older [43] |
| Elexacaftor–tezacaftor–ivacaftor (KAFTRIO/TRIKAFTA) (elexacaftor increases the number of CFTR proteins on the cell surface) | Indicated in patients who have at least one F508del mutation in the CFTR gene [35,36,37]. | First approval—2020 Currently approved for patients 6 years and older [37]. | First approval—2019 Currently approved for patients 2 years and older [35]. | First approval—2020 Currently approved for patients aged 2 years and older [36]. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
O’Regan, P.W.; Stevens, N.E.; Logan, N.; Ryan, D.J.; Maher, M.M. Paediatric Thoracic Imaging in Cystic Fibrosis in the Era of Cystic Fibrosis Transmembrane Conductance Regulator Modulation. Children 2024, 11, 256. https://doi.org/10.3390/children11020256
O’Regan PW, Stevens NE, Logan N, Ryan DJ, Maher MM. Paediatric Thoracic Imaging in Cystic Fibrosis in the Era of Cystic Fibrosis Transmembrane Conductance Regulator Modulation. Children. 2024; 11(2):256. https://doi.org/10.3390/children11020256
Chicago/Turabian StyleO’Regan, Patrick W., Niamh E. Stevens, Niamh Logan, David J. Ryan, and Michael M. Maher. 2024. "Paediatric Thoracic Imaging in Cystic Fibrosis in the Era of Cystic Fibrosis Transmembrane Conductance Regulator Modulation" Children 11, no. 2: 256. https://doi.org/10.3390/children11020256