
Genetic Disruption of Cilia-Associated Signaling Pathways in Patients with VACTERL Association
 , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Patients and Basic Data Set
2.2. Analysis of Protein Variation Effect and Gene Function
2.3. Sanger Sequencing
2.4. Literature Research and Illustrations
3. Results
3.1. Whole-Exome Sequencing
3.2. Signaling Pathways
3.2.1. Sonic Hedgehog Pathway
3.2.2. Wingless/Int-1 Pathway
3.2.3. Wnt and Shh Pathways
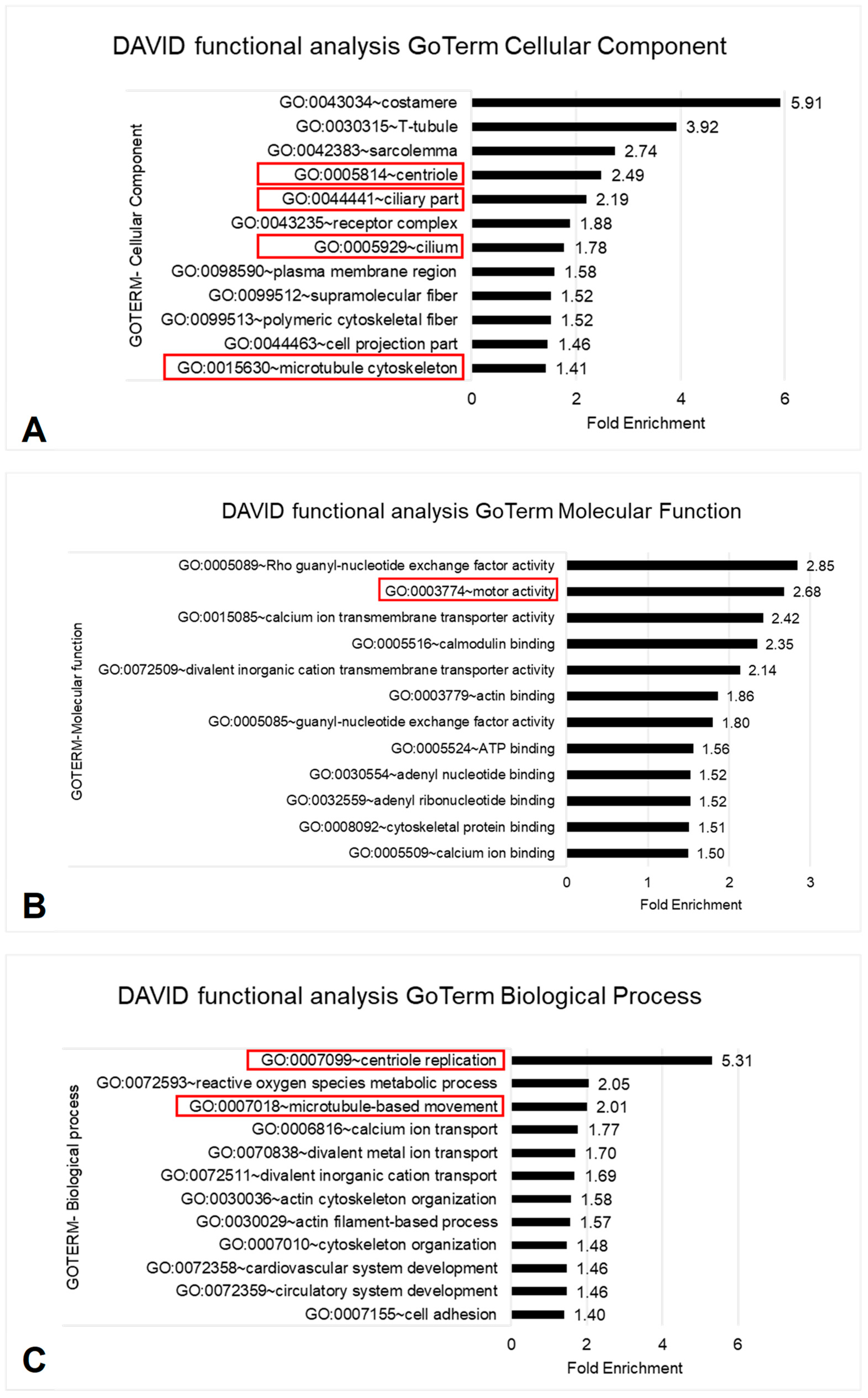
3.3. Functional Enrichment Analysis
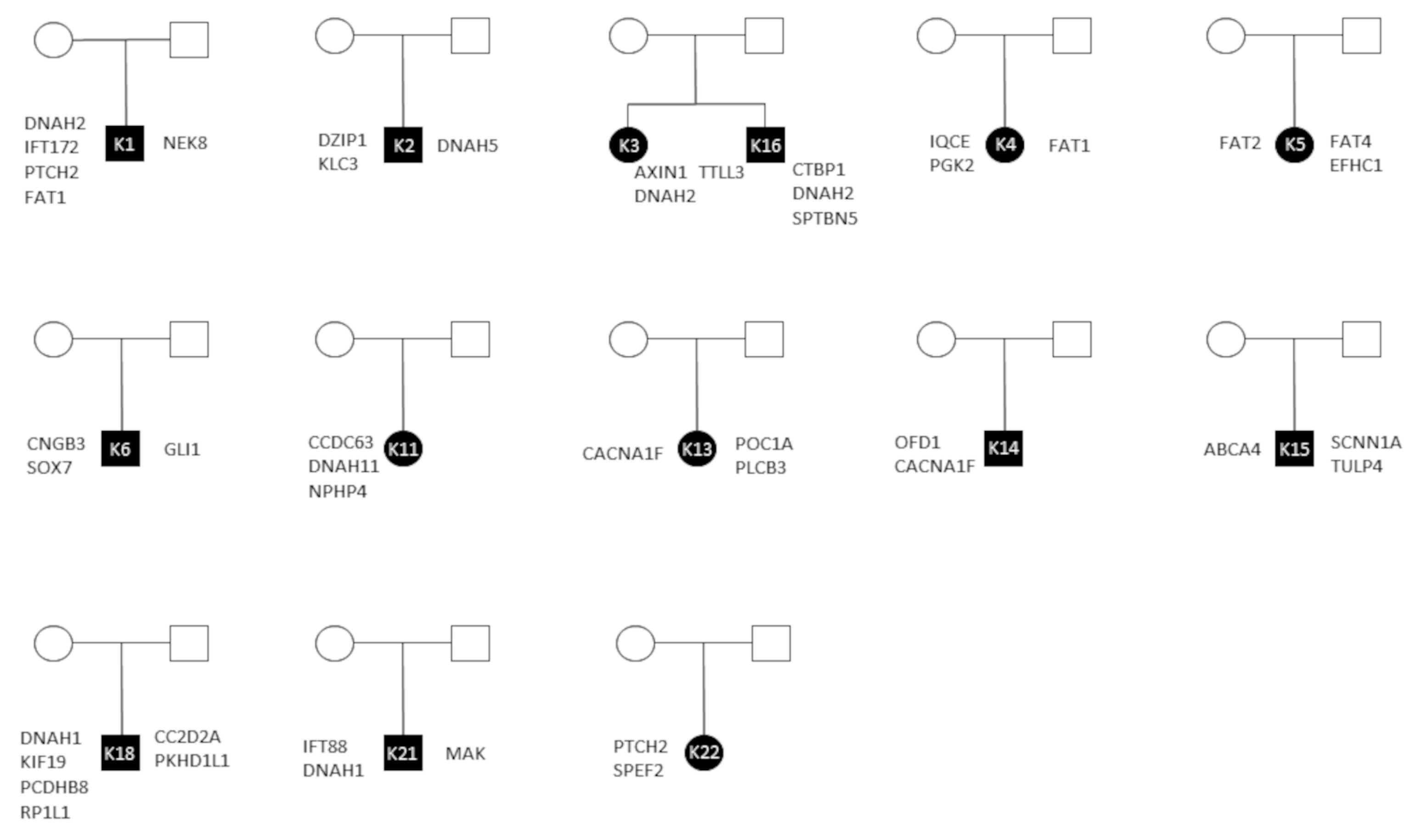
3.4. Inheritance
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Quan, L.; Smith, D.W. The VATER association. J. Pediatrics 1973, 82, 104–107. [Google Scholar] [CrossRef]
- Temtamy, S.A.; Miller, J.D. Extending the scope of the VATER association: Definition of the VATER syndrome. J. Pediatr. 1974, 85, 345–349. [Google Scholar] [CrossRef]
- Nora, A.H.; Nora, J.J. A syndrome of multiple congenital anomalies associated with teratogenic exposure. Arch. Environ. Health 1975, 30, 17–21. [Google Scholar] [CrossRef] [PubMed]
- Khoury, M.J.; Cordero, J.F.; Greenberg, F.; James, L.M.; Erickson, J.D. A population study of the VACTERL association: Evidence for its etiologic heterogeneity. Pediatrics 1983, 71, 815–820. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Liu, Z.; Chen, J.; Zuo, Y.; Liu, S.; Chen, W.; Liu, G.; Qiu, G.; Giampietro, P.F.; Wu, N.; et al. The genetic landscape and clinical implications of vertebral anomalies in VACTERL association. J. Med. Genet. 2016, 53, 431–437. [Google Scholar] [CrossRef]
- Stevenson, R.E.; Hunter, A.G.W. Considering the Embryopathogenesis of VACTERL Association. Mol. Syndromol. 2013, 4, 7–15. [Google Scholar] [CrossRef]
- Hilger, A.C.; Halbritter, J.; Pennimpede, T.; van der Ven, A.; Sarma, G.; Braun, D.A.; Porath, J.D.; Kohl, S.; Hwang, D.-Y.; Dworschak, G.C.; et al. Targeted Resequencing of 29 Candidate Genes and Mouse Expression Studies Implicate ZIC3 and FOXF1 in Human VATER/VACTERL Association. Hum. Mutat. 2015, 36, 1150–1154. [Google Scholar] [CrossRef] [PubMed]
- Kause, F.; Zhang, R.; Ludwig, M.; Schmiedeke, E.; Rissmann, A.; Thiele, H.; Altmueller, J.; Herms, S.; Hilger, A.C.; Hildebrandt, F.; et al. HSPA6: A new autosomal recessive candidate gene for the VATER/VACTERL malformation spectrum. Birth Defects Res. 2019, 111, 591–597. [Google Scholar] [CrossRef]
- Moreno, O.M.; Sánchez, A.I.; Herreño, A.; Giraldo, G.; Suárez, F.; Prieto, J.C.; Clavijo, A.S.; Olaya, M.; Vargas, Y.; Benítez, J.; et al. Phenotypic Characteristics and Copy Number Variants in a Cohort of Colombian Patients with VACTERL Association. Mol. Syndromol. 2020, 11, 271–283. [Google Scholar] [CrossRef]
- Stevens, S.J.C.; Stumpel, C.T.R.M.; Diderich, K.E.M.; van Slegtenhorst, M.A.; Abbott, M.-A.; Manning, C.; Balciuniene, J.; Pyle, L.C.; Leonard, J.; Murrell, J.R.; et al. The broader phenotypic spectrum of congenital caudal abnormalities associated with mutations in the caudal type homeobox 2 gene. Clin. Genet. 2021, 10, 183–189. [Google Scholar] [CrossRef]
- Kolvenbach, C.M.; van der Ven, A.T.; Kause, F.; Shril, S.; Scala, M.; Connaughton, D.M.; Mann, N.; Nakayama, M.; Dai, R.; Kitzler, T.M.; et al. Exome survey of individuals affected by VATER/VACTERL with renal phenotypes identifies phenocopies and novel candidate genes. Am. J. Med. Genet. Part A 2021, 185, 3784–3792. [Google Scholar] [CrossRef] [PubMed]
- Martin, E.M.M.A.; Enriquez, A.; Sparrow, D.B.; Humphreys, D.T.; McInerney-Leo, A.M.; Leo, P.J.; Duncan, E.L.; Iyer, K.R.; Greasby, J.A.; Ip, E.; et al. Heterozygous loss of WBP11 function causes multiple congenital defects in humans and mice. Hum. Mol. Genet. 2020, 29, 3662–3678. [Google Scholar] [CrossRef] [PubMed]
- Kim, P.C.; Mo, R.; Hui Cc, C. Murine models of VACTERL syndrome: Role of sonic hedgehog signaling pathway. J. Pediatr. Surg. 2001, 36, 381–384. [Google Scholar] [CrossRef] [PubMed]
- Friedland-Little, J.M.; Hoffmann, A.D.; Ocbina, P.J.R.; Peterson, M.A.; Bosman, J.D.; Chen, Y.; Cheng, S.Y.; Anderson, K.V.; Moskowitz, I.P. A novel murine allele of Intraflagellar Transport Protein 172 causes a syndrome including VACTERL-like features with hydrocephalus. Hum. Mol. Genet. 2011, 20, 3725–3737. [Google Scholar] [CrossRef]
- Taschner, M.; Lorentzen, E. The Intraflagellar Transport Machinery. Cold Spring Harb. Perspect. Biol. 2016, 8, a028092. [Google Scholar] [CrossRef]
- Wheway, G.; Nazlamova, L.; Hancock, J.T. Signaling through the Primary Cilium. Front. Cell Dev. Biol. 2018, 6, 8. [Google Scholar] [CrossRef]
- Yuan, X.; Yang, S. Cilia/Ift protein and motor -related bone diseases and mouse models. Front. Biosci. (Landmark Ed.) 2015, 20, 515–555. [Google Scholar] [CrossRef]
- Satir, P.; Christensen, S.T. Overview of structure and function of mammalian cilia. Annu. Rev. Physiol. 2007, 69, 377–400. [Google Scholar] [CrossRef]
- Gabriel, G.C.; Pazour, G.J.; Lo, C.W. Congenital Heart Defects and Ciliopathies Associated With Renal Phenotypes. Front. Pediatr. 2018, 6, 175. [Google Scholar] [CrossRef]
- Ibañez-Tallon, I.; Heintz, N.; Omran, H. To beat or not to beat: Roles of cilia in development and disease. Hum. Mol. Genetics. 2003, 12 Spec No 1, R27–R35. [Google Scholar] [CrossRef]
- Verhey, K.J.; Dishinger, J.; Kee, H.L. Kinesin motors and primary cilia. Biochem. Soc. Trans. 2011, 39, 1120–1125. [Google Scholar] [CrossRef]
- Goetz, S.C.; Anderson, K.V. The primary cilium: A signalling centre during vertebrate development. Nat. Rev. Genet. 2010, 11, 331–344. [Google Scholar] [CrossRef] [PubMed]
- Halbritter, J.; Bizet, A.A.; Schmidts, M.; Porath, J.D.; Braun, D.A.; Gee, H.Y.; McInerney-Leo, A.M.; Krug, P.; Filhol, E.; Davis, E.E.; et al. Defects in the IFT-B component IFT172 cause Jeune and Mainzer-Saldino syndromes in humans. Am. J. Hum. Genet. 2013, 93, 915–925. [Google Scholar] [CrossRef] [PubMed]
- Kim, A.; Savary, C.; Dubourg, C.; Carré, W.; Mouden, C.; Hamdi-Rozé, H.; Guyodo, H.; Le Douce, J.; Pasquier, L.; Flori, E.; et al. Integrated clinical and omics approach to rare diseases: Novel genes and oligogenic inheritance in holoprosencephaly. Brain 2019, 142, 35–49. [Google Scholar] [CrossRef]
- Ritter, J.; Lisec, K.; Heinrich, M.; von Schweinitz, D.; Kappler, R.; Hubertus, J. Genetic Evidence for Congenital Vascular Disorders in Patients with VACTERL Association. Eur. J. Pediatr. Surg. Off. J. Austrian Assoc. Pediatr. Surg. 2022, 32, 61–66. [Google Scholar] [CrossRef] [PubMed]
- Adzhubei, I.A.; Schmidt, S.; Peshkin, L.; Ramensky, V.E.; Gerasimova, A.; Bork, P.; Kondrashov, A.S.; Sunyaev, S.R. A method and server for predicting damaging missense mutations. Nat. Methods 2010, 7, 248–249. [Google Scholar] [CrossRef]
- Choi, Y.; Chan, A.P. PROVEAN web server: A tool to predict the functional effect of amino acid substitutions and indels. Bioinformatics 2015, 31, 2745–2747. [Google Scholar] [CrossRef]
- Choi, Y. A fast computation of pairwise sequence alignment scores between a protein and a set of single-locus variants of another protein. In Proceedings of the ACM Conference on Bioinformatics, Computational Biology and Biomedicine, Orlando, FL, USA, 7–10 October 2012; Ranka, S., Ed.; ACM: New York, NY, USA, 2012; pp. 414–417, ISBN 9781450316705. [Google Scholar]
- Choi, Y.; Sims, G.E.; Murphy, S.; Miller, J.R.; Chan, A.P. Predicting the functional effect of amino acid substitutions and indels. PLoS ONE 2012, 7, e46688. [Google Scholar] [CrossRef]
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alföldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; Birnbaum, D.P.; et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 2020, 581, 434–443. [Google Scholar] [CrossRef]
- Yoshimura, K.; Hanaoka, T.; Ohnami, S.; Ohnami, S.; Kohno, T.; Liu, Y.; Yoshida, T.; Sakamoto, H.; Tsugane, S. Allele frequencies of single nucleotide polymorphisms (SNPs) in 40 candidate genes for gene-environment studies on cancer: Data from population-based Japanese random samples. J. Hum. Genet. 2003, 48, 654–658. [Google Scholar] [CrossRef]
- Bamshad, M.J.; Ng, S.B.; Bigham, A.W.; Tabor, H.K.; Emond, M.J.; Nickerson, D.A.; Shendure, J. Exome sequencing as a tool for Mendelian disease gene discovery. Nat. Rev. Genet. 2011, 12, 745–755. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef] [PubMed]
- Jassal, B.; Matthews, L.; Viteri, G.; Gong, C.; Lorente, P.; Fabregat, A.; Sidiropoulos, K.; Cook, J.; Gillespie, M.; Haw, R.; et al. The reactome pathway knowledgebase. Nucleic Acids Res. 2020, 48, D498–D503. [Google Scholar] [CrossRef] [PubMed]
- Kimelman, D. Signaling by Wnt. Reactome 2007. [Google Scholar] [CrossRef]
- Rothfels, K. Hedgehog ligand biogenesis. Reactome 2014. [Google Scholar] [CrossRef]
- Rothfels, K. Hedgehog ‘off’ State. Reactome 2014. [Google Scholar] [CrossRef]
- Rothfels, K. Hedgehog ‘on’ state. Reactome 2014. [Google Scholar] [CrossRef]
- Safran, M.; Dalah, I.; Alexander, J.; Rosen, N.; Iny Stein, T.; Shmoish, M.; Nativ, N.; Bahir, I.; Doniger, T.; Krug, H.; et al. GeneCards Version 3: The human gene integrator. Database 2010, 2010, baq020. [Google Scholar] [CrossRef]
- UniProt: The universal protein knowledgebase in 2021. Nucleic Acids Res. 2021, 49, D480–D489. [CrossRef]
- Sayers, E.W.; Bolton, E.E.; Brister, J.R.; Canese, K.; Chan, J.; Comeau, D.C.; Connor, R.; Funk, K.; Kelly, C.; Kim, S.; et al. Database resources of the national center for biotechnology information. Nucleic Acids Res. 2022, 50, D20–D26. [Google Scholar] [CrossRef]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Bioinformatics enrichment tools: Paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009, 37, e16. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef] [PubMed]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene ontology: Tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [PubMed]
- The Gene Ontology resource: Enriching a GOld mine. Nucleic Acids Res. 2021, 49, D325–D334. [CrossRef]
- Ye, J.; Coulouris, G.; Zaretskaya, I.; Cutcutache, I.; Rozen, S.; Madden, T.L. Primer-BLAST: A tool to design target-specific primers for polymerase chain reaction. BMC Bioinform. 2012, 13, 134. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Kim, P.; Hui, C.C. The VACTERL association: Lessons from the Sonic hedgehog pathway. Clinical Genetics 2001, 59, 306–315. [Google Scholar] [CrossRef] [PubMed]
- Ingham, P.W.; McMahon, A.P. Hedgehog signaling in animal development: Paradigms and principles. Genes Dev. 2001, 15, 3059–3087. [Google Scholar] [CrossRef]
- Saitsu, H.; Sonoda, M.; Higashijima, T.; Shirozu, H.; Masuda, H.; Tohyama, J.; Kato, M.; Nakashima, M.; Tsurusaki, Y.; Mizuguchi, T.; et al. Somatic mutations in GLI3 and OFD1 involved in sonic hedgehog signaling cause hypothalamic hamartoma. Ann. Clin. Transl. Neurol. 2016, 3, 356–365. [Google Scholar] [CrossRef]
- Gallardo, V.; Bovolenta, P. Positive and negative regulation of Shh signalling in vertebrate retinal development. F1000Research 2018, 7, 1934. [Google Scholar] [CrossRef]
- Skoda, A.M.; Simovic, D.; Karin, V.; Kardum, V.; Vranic, S.; Serman, L. The role of the Hedgehog signaling pathway in cancer: A comprehensive review. Bosn. J. Basic Med. Sci. 2018, 18, 8–20. [Google Scholar] [CrossRef]
- Umair, M.; Shah, K.; Alhaddad, B.; Haack, T.B.; Graf, E.; Strom, T.M.; Meitinger, T.; Ahmad, W. Exome sequencing revealed a splice site variant in the IQCE gene underlying post-axial polydactyly type A restricted to lower limb. Eur. J. Hum. Genet. 2017, 25, 960–965. [Google Scholar] [CrossRef] [PubMed]
- Jin, Z.; Mei, W.; Strack, S.; Jia, J.; Yang, J. The antagonistic action of B56-containing protein phosphatase 2As and casein kinase 2 controls the phosphorylation and Gli turnover function of Daz interacting protein 1. J. Biol. Chem. 2011, 286, 36171–36179. [Google Scholar] [CrossRef] [PubMed]
- Huangfu, D.; Anderson, K.V. Cilia and Hedgehog responsiveness in the mouse. Proc. Natl. Acad. Sci. USA. 2005, 102, 11325–11330. [Google Scholar] [CrossRef] [PubMed]
- Villavicencio, E.H.; Walterhouse, D.O.; Iannaccone, P.M. The Sonic Hedgehog–Patched–Gli Pathway in Human Development and Disease. Am. J. Hum. Genet. 2000, 67, 1047–1054. [Google Scholar] [CrossRef] [PubMed]
- Cai, J.; Feng, D.; Hu, L.; Chen, H.; Yang, G.; Cai, Q.; Gao, C.; Wei, D. FAT4 functions as a tumour suppressor in gastric cancer by modulating Wnt/β-catenin signalling. Br. J. Cancer 2015, 113, 1720–1729. [Google Scholar] [CrossRef]
- Huard, C.C.; Tremblay, C.S.; Helsper, K.; Delisle, M.-C.; Schindler, D.; Lévesque, G.; Carreau, M. Fanconi anemia proteins interact with CtBP1 and modulate the expression of the Wnt antagonist Dickkopf-1. Blood 2013, 121, 1729–1739. [Google Scholar] [CrossRef]
- Morris, L.G.T.; Kaufman, A.M.; Gong, Y.; Ramaswami, D.; Walsh, L.A.; Turcan, Ş.; Eng, S.; Kannan, K.; Zou, Y.; Peng, L.; et al. Recurrent somatic mutation of FAT1 in multiple human cancers leads to aberrant Wnt activation. Nat. Genet. 2013, 45, 253–261. [Google Scholar] [CrossRef]
- Arce, L.; Pate, K.T.; Waterman, M.L. Groucho binds two conserved regions of LEF-1 for HDAC-dependent repression. BMC Cancer 2009, 9, 159. [Google Scholar] [CrossRef]
- Ellwanger, K.; Saito, H.; Clément-Lacroix, P.; Maltry, N.; Niedermeyer, J.; Lee, W.K.; Baron, R.; Rawadi, G.; Westphal, H.; Niehrs, C. Targeted disruption of the Wnt regulator Kremen induces limb defects and high bone density. Mol. Cell. Biol. 2008, 28, 4875–4882. [Google Scholar] [CrossRef]
- Guo, L.; Zhong, D.; Lau, S.; Liu, X.; Dong, X.-Y.; Sun, X.; Yang, V.W.; Vertino, P.M.; Moreno, C.S.; Varma, V.; et al. Sox7 Is an independent checkpoint for beta-catenin function in prostate and colon epithelial cells. Mol. Cancer Res. 2008, 6, 1421–1430. [Google Scholar] [CrossRef]
- Kusano, S.; Raab-Traub, N. I-mfa domain proteins interact with Axin and affect its regulation of the Wnt and c-Jun N-terminal kinase signaling pathways. Mol. Cell. Biol. 2002, 22, 6393–6405. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, H.; Murata, Y.; Koyama, K.; Fujiyama, A.; Miyoshi, Y.; Monden, M.; Akiyama, T.; Nakamura, Y. Identification of a brain-specific APC homologue, APCL, and its interaction with beta-catenin. Cancer Res. 1998, 58, 5176–5181. [Google Scholar]
- Mao, J.; Wang, J.; Liu, B.; Pan, W.; Farr, G.H.; Flynn, C.; Yuan, H.; Takada, S.; Kimelman, D.; Li, L.; et al. Low-Density Lipoprotein Receptor-Related Protein-5 Binds to Axin and Regulates the Canonical Wnt Signaling Pathway. Mol. Cell 2001, 7, 801–809. [Google Scholar] [CrossRef] [PubMed]
- Schneider, A.J.; Branam, A.M.; Peterson, R.E. Intersection of AHR and Wnt signaling in development, health, and disease. Int. J. Mol. Sci. 2014, 15, 17852–17885. [Google Scholar] [CrossRef]
- Zhang, X.; Liu, J.; Liang, X.; Chen, J.; Hong, J.; Li, L.; He, Q.; Cai, X. History and progression of Fat cadherins in health and disease. Onco. Targets. Ther. 2016, 9, 7337–7343. [Google Scholar] [CrossRef] [PubMed]
- Grigoryan, T.; Wend, P.; Klaus, A.; Birchmeier, W. Deciphering the function of canonical Wnt signals in development and disease: Conditional loss- and gain-of-function mutations of beta-catenin in mice. Genes Dev. 2008, 22, 2308–2341. [Google Scholar] [CrossRef]
- Miyagawa, S.; Harada, M.; Matsumaru, D.; Tanaka, K.; Inoue, C.; Nakahara, C.; Haraguchi, R.; Matsushita, S.; Suzuki, K.; Nakagata, N.; et al. Disruption of the temporally regulated cloaca endodermal β-catenin signaling causes anorectal malformations. Cell Death Differ. 2014, 21, 990–997. [Google Scholar] [CrossRef]
- Alfaro, A.C.; Roberts, B.; Kwong, L.; Bijlsma, M.F.; Roelink, H. Ptch2 mediates the Shh response in Ptch1-/- cells. Development 2014, 141, 3331–3339. [Google Scholar] [CrossRef]
- Bartoloni, L.; Blouin, J.-L.; Pan, Y.; Gehrig, C.; Maiti, A.K.; Scamuffa, N.; Rossier, C.; Jorissen, M.; Armengot, M.; Meeks, M.; et al. Mutations in the DNAH11 (axonemal heavy chain dynein type 11) gene cause one form of situs inversus totalis and most likely primary ciliary dyskinesia. Proc. Natl. Acad. Sci. USA. 2002, 99, 10282–10286. [Google Scholar] [CrossRef]
- Imtiaz, F.; Allam, R.; Ramzan, K.; Al-Sayed, M. Variation in DNAH1 may contribute to primary ciliary dyskinesia. BMC Med. Genet. 2015, 16, 14. [Google Scholar] [CrossRef]
- Zhang, Y.; Chen, Y.; Zheng, J.; Wang, J.; Duan, S.; Zhang, W.; Yan, X.; Zhu, X. Vertebrate Dynein-f depends on Wdr78 for axonemal localization and is essential for ciliary beat. J. Mol. Cell Biol. 2019, 11, 383–394. [Google Scholar] [CrossRef]
- Zukas, R.; Chang, A.J.; Rice, M.; Springer, A.L. Structural analysis of flagellar axonemes from inner arm dynein knockdown strains of Trypanosoma brucei. Biocell 2012, 36, 133–141. [Google Scholar] [CrossRef]
- Olbrich, H.; Häffner, K.; Kispert, A.; Völkel, A.; Volz, A.; Sasmaz, G.; Reinhardt, R.; Hennig, S.; Lehrach, H.; Konietzko, N.; et al. Mutations in DNAH5 cause primary ciliary dyskinesia and randomization of left-right asymmetry. Nat. Genet. 2002, 30, 143–144. [Google Scholar] [CrossRef]
- Liu, S.; Chen, W.; Zhan, Y.; Li, S.; Ma, X.; Ma, D.; Sheng, W.; Huang, G. DNAH11 variants and its association with congenital heart disease and heterotaxy syndrome. Sci. Rep. 2019, 9, 6683. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| All | Wnt-Group | Shh-Group | Wnt/Shh-Group | DNAH-Group | IFT-Group | |
|---|---|---|---|---|---|---|
| n (%) | n (%) | n (%) | n (%) | n (%) | n (%) | |
| Total | 21 (100) | 13 (62) | 10 (48) | 6 (29) | 9 (43) | 5 (24) |
| Gender | ||||||
| Female | 8 (38) | 5 (24) | 3 (14) | 2 (10) | 2 (10) | 1 (5) |
| Male | 13 (62) | 8 (38) | 7 (33) | 4 (19) | 7 (33) | 4 (19) |
| Female-to-male ratio | 1:1.6 | 1:1.6 | 1:2.3 | 1:2.0 | 1:3.5 | 1:4.0 |
| Age at the end of data acquisition (December 2019) Years, median (range) | 10.8 (1–31) | 11.1 (1–31) | 9.3 (2–19) | 9.7 (3–19) | 8.2 (1–16) | 11.2 (5–17) |
| Fulfilled VACTERL criteria | ||||||
| 2 | 1 (5) | 0 (0) | 1 (5) | 0 (0) | 1 (5) | 1 (5) |
| 3 | 7 (33) | 4 (19) | 5 (24) | 3 (14) | 4 (19) | 2 (10) |
| 4 | 4 (19) | 4 (19) | 0 (0) | 0 (0) | 2 (10) | 0 (0) |
| 5 | 8 (38) | 5 (24) | 3 (14) | 3 (14) | 1 (5) | 2 (10) |
| 6 | 1 (5) | 0 (0) | 1 (5) | 0 (0) | 1 (5) | 0 (0) |
| Distribution of VACTERL criteria | ||||||
| Vertebral anomalies | 17 (81) | 10 (48) | 7 (33) | 4 (19) | 6 (29) | 3 (14) |
| Anorectal malformation | 12 (57) | 8 (38) | 6 (29) | 5 (24) | 5 (24) | 3 (14) |
| Cardiac defects | 17 (81) | 12 (57) | 7 (33) | 5 (24) | 7 (33) | 3 (14) |
| Tracheoesophageal defects | 16 (76) | 10 (48) | 9 (43) | 5 (24) | 6 (29) | 5 (24) |
| Renal anomalies | 15 (71) | 8 (38) | 7 (33) | 4 (19) | 7 (33) | 4 (19) |
| Limb anomalies | 8 (38) | 5 (24) | 2 (10) | 1 (5) | 2 (10) | 0 (0) |
| Distribution of other anomalies | ||||||
| Other gastro-intestinal anomalies | 9 (43) | 5 (24) | 5 (24) | 3 (14) | 3 (14) | 1 (5) |
| Oro-facio-auricolo anomalies | 8 (38) | 4 (19) | 4 (19) | 1 (5) | 3 (14) | 1 (5) |
| Genital anomalies | 8 (38) | 4 (19) | 4 (19) | 2 (10) | 6 (29) | 3 (14) |
| Patient | Gender | Fulfilled VACTERL Criteria | Shh [49,50,51,52,53,54,55] | Wnt [56,57,58,59,60,61,62,63,64] |
|---|---|---|---|---|
| VCK1 | M | V, A, C, TE, R | PTCH2, IFT172 | FAT1 |
| VCK2 | M | V, A, C, TE, R, L | DZIP1 | |
| VCK3 | F | V, A, C, R | AXIN1 | |
| VCK4 | F | V, A, C, TE, L | IQCE | FAT1 |
| VCK5 | F | V, A, C, TE, R | FAT2, FAT4 | |
| VCK6 | M | V, A, C, TE, R | GLI1 | SOX7 |
| VCK7 | F | V, A, C, R, L | ||
| VCK8 | M | V, A, C, TE, R | ||
| VCK9 | M | V, C, TE, R, L | ||
| VCK10 | M | V, C, TE, L | KREMEN2, AP2A2 | |
| VCK11 | F | V, A, R | ||
| VCK13 | F | V, C, TE, L | PLCB3 | |
| VCK14 | M | V, A, C | OFD1 | TLE5 |
| VCK15 | M | V, C, TE, R, L | GNAT2 | |
| VCK16 | M | V, A, C, R | CTBP1 | |
| VCK17 | F | A, TE, R | IFT57 | FAT4 |
| VCK18 | M | C, TE, L | LRP5 | |
| VCK19 | M | C, TE, R | LRP2 | |
| VCK20 | M | C, TE, R | CSNK1G1 | FAT4, APC2, MMP7 |
| VCK21 | M | V, TE | IFT88 | |
| VCK22 | F | V, TE, R | PTCH2 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ritter, J.; Lisec, K.; Klinner, M.; Heinrich, M.; von Schweinitz, D.; Kappler, R.; Hubertus, J. Genetic Disruption of Cilia-Associated Signaling Pathways in Patients with VACTERL Association. Children 2023, 10, 882. https://doi.org/10.3390/children10050882
Ritter J, Lisec K, Klinner M, Heinrich M, von Schweinitz D, Kappler R, Hubertus J. Genetic Disruption of Cilia-Associated Signaling Pathways in Patients with VACTERL Association. Children. 2023; 10(5):882. https://doi.org/10.3390/children10050882
Chicago/Turabian StyleRitter, Jessica, Kristina Lisec, Marina Klinner, Martina Heinrich, Dietrich von Schweinitz, Roland Kappler, and Jochen Hubertus. 2023. "Genetic Disruption of Cilia-Associated Signaling Pathways in Patients with VACTERL Association" Children 10, no. 5: 882. https://doi.org/10.3390/children10050882