
The Cutting Edge of Disease Modeling: Synergy of Induced Pluripotent Stem Cell Technology and Genetically Encoded Biosensors


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
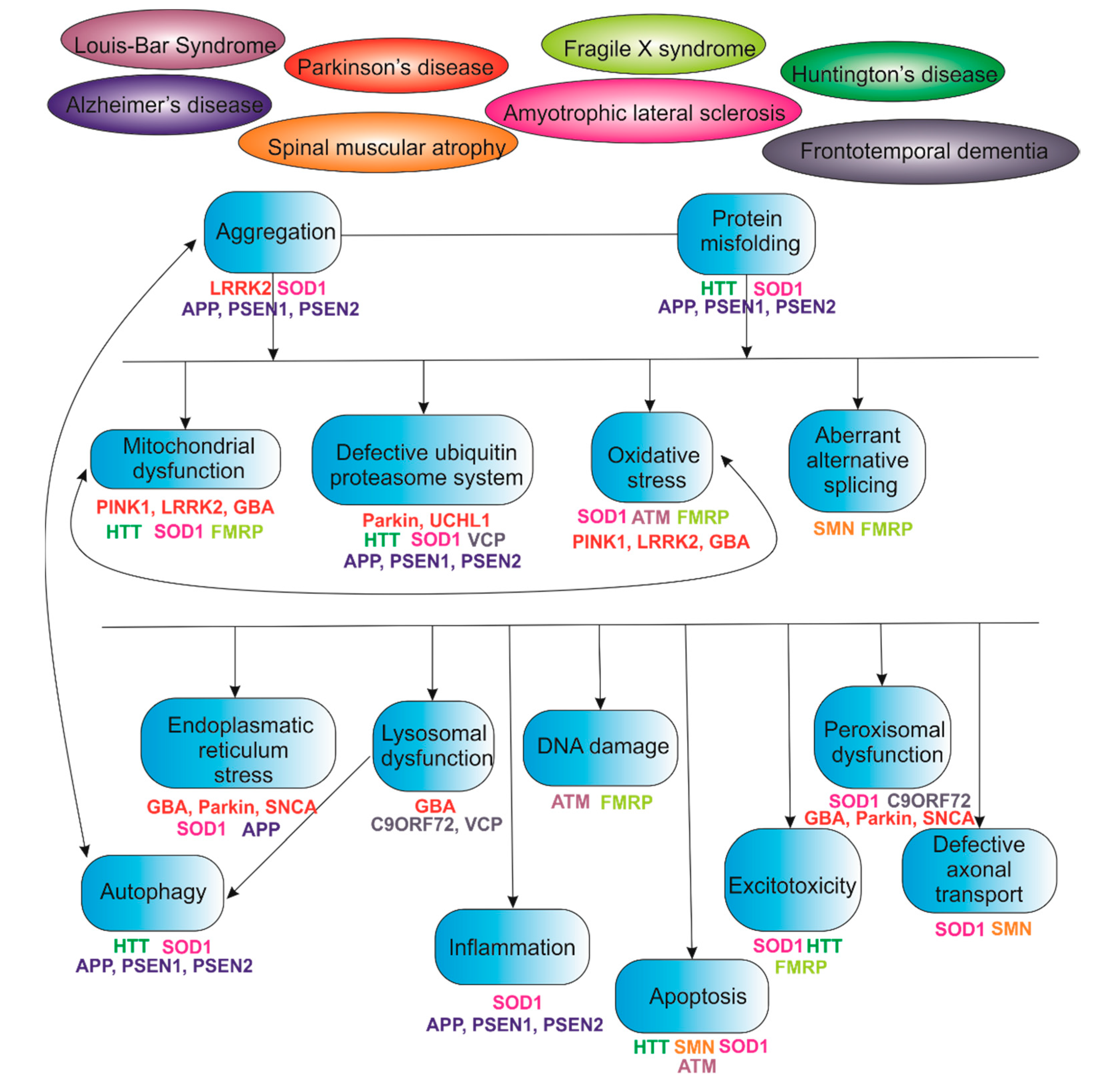
2. IPSCs and Cell Models
3. Biosensors Classification
4. Chemical Biosensors
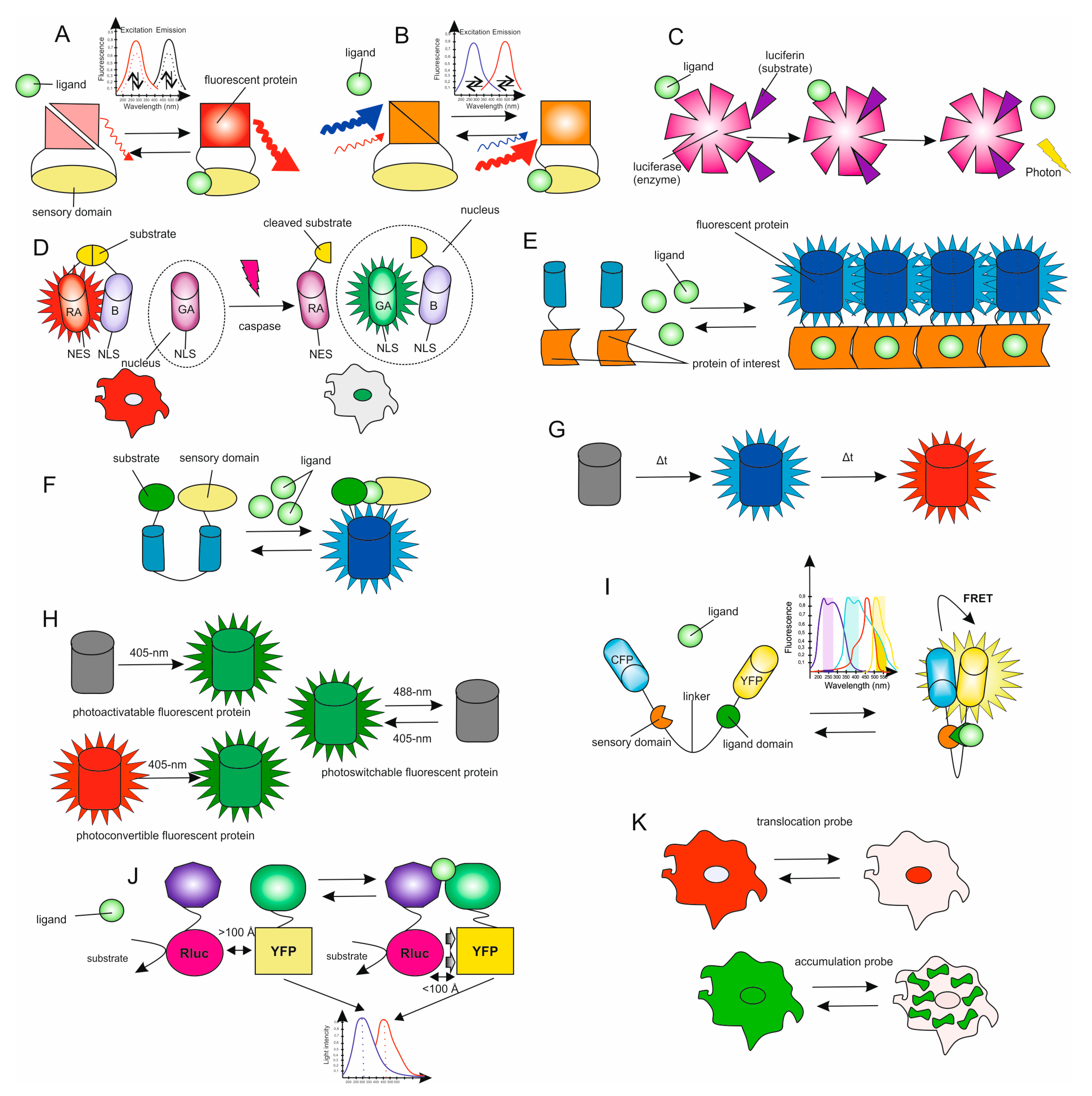
5. Genetically Encoded Biosensors
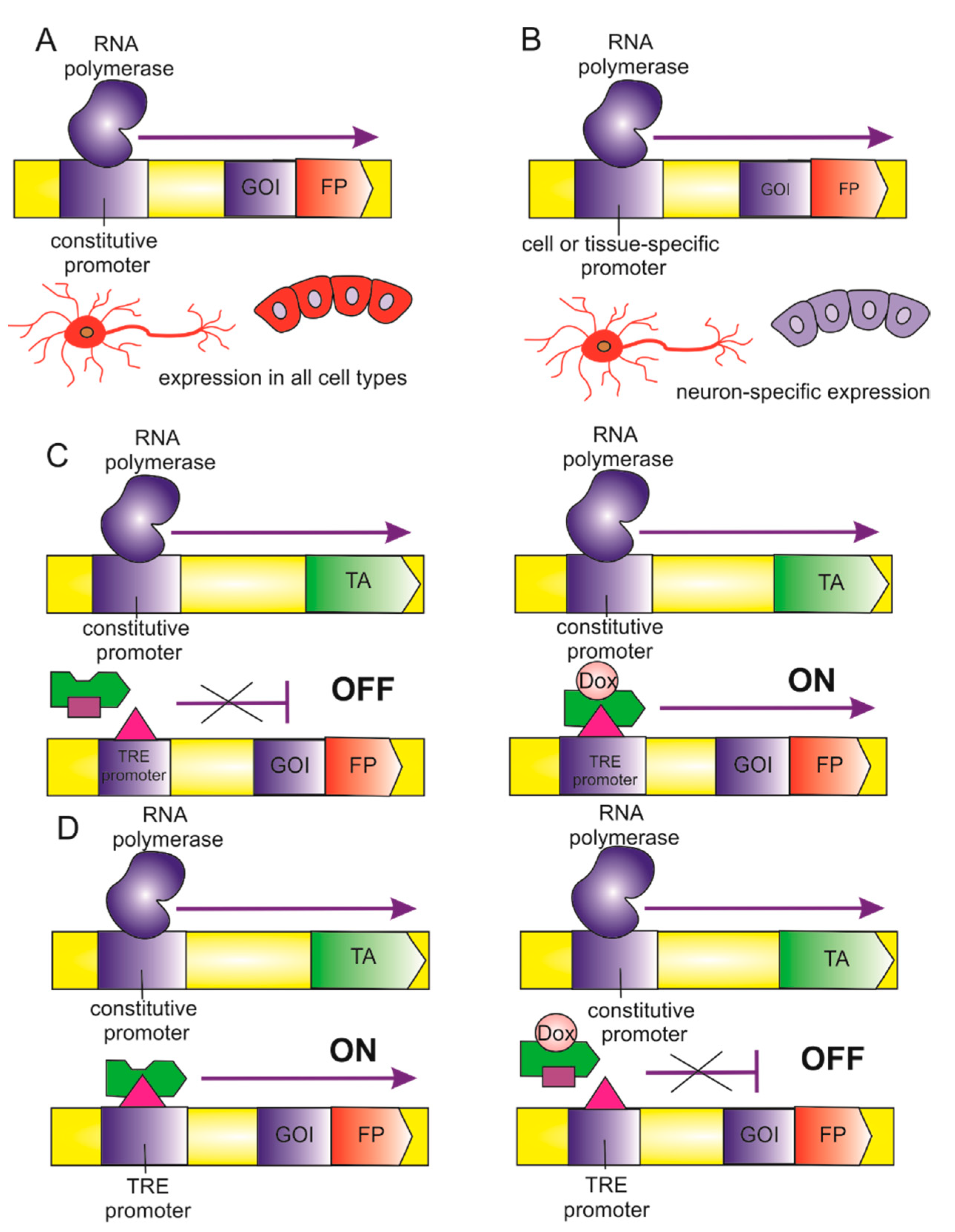
5.1. Biosensor Expression Regulation
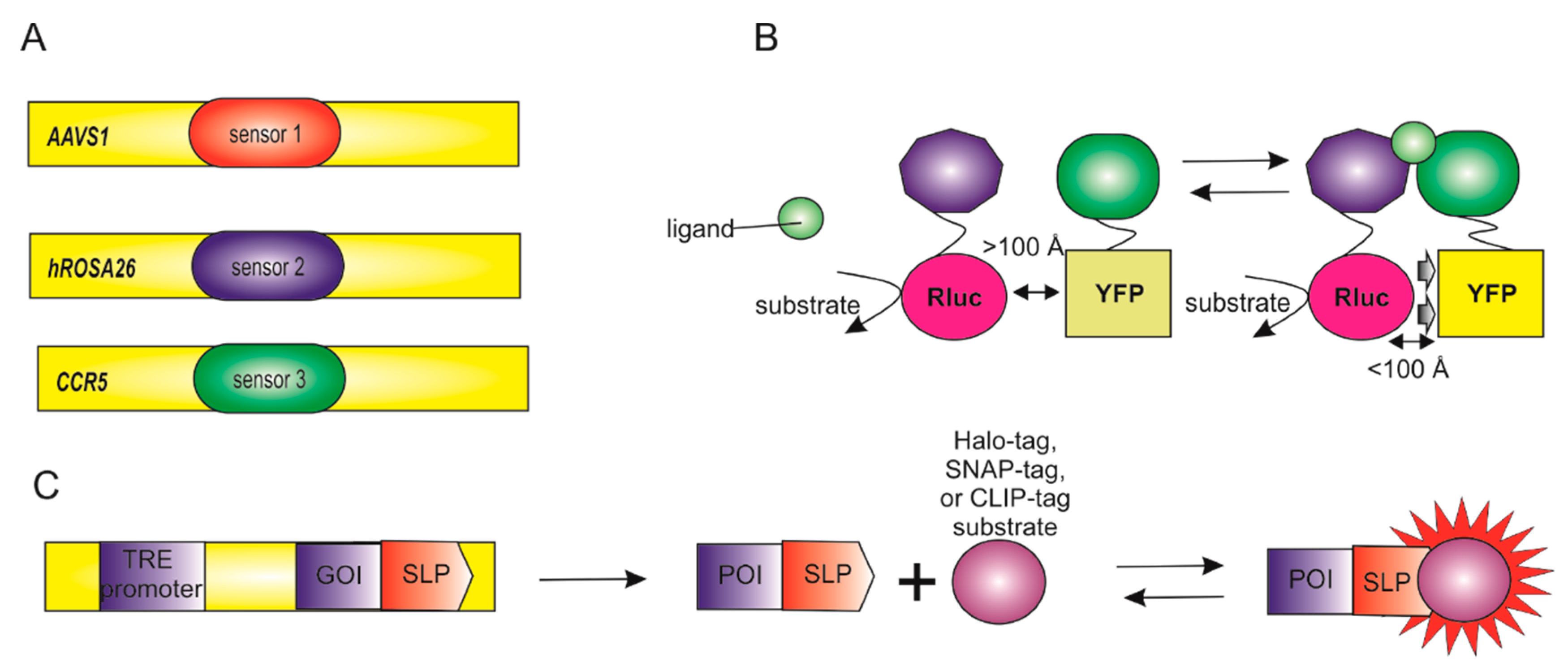
5.2. Combination of Biosensors
5.3. Biosensor Selection
- Ease of use. One of the critical factors when choosing a biosensor is the availability of the equipment needed for the measurements. For example, ratiometric biosensors based on FRET require sophisticated microscopic equipment to quickly (or simultaneously) collect data from two or more fluorescence channels. At the same time, for biosensors that measure fluorescence intensity, uninvolved instruments are needed to collect data. It is also necessary to consider the availability of the materials used. Chemically encoded biosensors must be constantly purchased, while genetically encoded sensors are infinitely renewable. In addition, some staining protocols using chemically encoded biosensors are cumbersome, especially if the biosensor is impermeable to cells. Finally, many luminescent biosensors require an additional step of adding a substrate such as coelenterazine [69].
- Consideration of the spectral features of the fluorophore. The spectral properties of the biosensors should be carefully analyzed to obtain two-color images and avoid crosstalk or fluorophore leaks; red-shifted excitation wavelengths are preferred for in vivo experiments due to their deep tissue permeability.
- Signal-to-noise ratio. This ratio depends on changes in fluorescence caused by ligand binding compared to fluctuations recorded by autofluorescence. To minimize photobleaching, researchers use the lowest excitation intensity and/or exposure time required to achieve an acceptable signal-to-noise ratio. For a 16-bit camera, a good rule of thumb is that the lowest signal is at least 1000 samples above the background.
- Sensitivity. One of the indicators of the sensitivity of a biosensor is the dynamic range, which is the ratio between the minimum and maximum values that the biosensor can detect. The sensitivity of the biosensor used is application specific. For example, to measure constant concentrations of a molecule or ion, it is recommended to choose a sensor with a dissociation constant (Kd) equal to or close to the expected concentration. An even more rigorous approach is to measure the concentration at rest using several biosensors, each of which has a slightly different Kd value. If the goal is to measure a change in concentration or activity when the signal is expected to be weak, then the biosensor with the highest dynamic range and Kd value, at which the concentration change will be within 5 Kd, will be most sensitive.
- Kinetics. The rate of binding/detachment of the ligand with the biosensor is crucial, which determines the ability or inability of the sensor to report rapid kinetics, as well as selectivity for the ligand, that is, the specificity of ligand binding in comparison with other molecules, and the sensor’s affinity for the ligand. The in situ environment can significantly affect the Kd and hence any quantitative analysis in living cells. This problem applies to any sensor and, in particular, to target sensors that are localized in subcellular niches, the environment of which can vary in terms of pH, viscosity, the concentration of heavy metals, and binding to local proteins. Moreover, the addition of the target peptide itself can affect the properties of the sensor. Therefore, accurate measurement of the Kd value is extremely important if any quantification is needed. As mentioned above, ratiometric sensors are easier to calibrate than sensors that only change signal strength. In the latter case, the information is usually qualitative rather than quantitative. Since cellular signals occur at different time scales, the kinetics of the process under study will also determine the optimal sensor to use. Small molecules often offer higher on and off rates (kon and koff) and thus give better resolution for events that occur on fast time scales. As a general rule of thumb, the kinetics of the sensor must accurately reproduce the biological signal. However, the slow turn-on may have some advantages in detecting low, weak signals.
- Signal localization. Genetically encoded sensors can easily target specific organelles or cell compartments by triggering localization signals. In the case of using chemical sensors, the task becomes more complicated. It is crucial to understand that the environment (pH, redox state) of a given organelle can limit the available sensors. For example, endocytic vesicles maintain an acidic pH (<6), which quenches YFP fluorescence and thus inhibits the use of CFP-YFP sensors in such acidic compartments.
- Quantification. For events requiring quantification, ratiometric biosensors or FRET sensors are generally preferred over fluorescence intensity sensors. FRET-based biosensors usually have a wider dynamic range. However, the fluorescence intensity is largely determined by the target molecule itself, as well as the level of sensor expression in a cell or organelle. Consequently, this type of biosensor is poorly suited for quantifying the concentration of an investigated molecule or changes in concentration. Calibration is needed to perform a comparative analysis of the data obtained using a particular biosensor in different types of cells or under the influence of various stimuli. It is necessary to calibrate the biosensor to measure the minimum and maximum signal levels. Although individual signal levels may vary from one cell to the next, the overall dynamic range must remain unchanged. It is also critical to be sure that the sensor response does not depend on the amount expressed in the cell. There should not be a strong correlation between the response and the concentration of the sensor because a strong correlation can lead to misinterpretation of the data obtained. This is especially true for chemical sensors, while the expression of genetically encoded sensors is controlled in various ways.
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Takahashi, K.; Yamanaka, S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef] [Green Version]
- Shefner, J.M.; Cudkowicz, M.E.; Schoenfeld, D.; Conrad, T.; Taft, J.; Chilton, M.; Urbinelli, L.; Qureshi, M.; Zhang, H.; Pestronk, A.; et al. A clinical trial of creatine in ALS. Neurology 2004, 63, 1656–1661. [Google Scholar] [CrossRef] [PubMed]
- Singh, B.; Gupta, R.S. Species-specific differences in the toxicity and mutagenicity of the anticancer drugs mithramycin, chromomycin A3, and olivomycin. Cancer Res. 1985, 45, 2813–2820. [Google Scholar]
- Studer, L.; Vera, E.; Cornacchia, D. Programming and Reprogramming Cellular Age in the Era of Induced Pluripotency. Cell Stem Cell 2015, 16, 591–600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, H.N.; Byers, B.; Cord, B.; Shcheglovitov, A.; Byrne, J.; Gujar, P.; Kee, K.; Schule, B.; Dolmetsch, R.E.; Langston, W.; et al. LRRK2 mutant iPSC-derived DA neurons demonstrate increased susceptibility to oxidative stress. Cell Stem Cell 2011, 8, 267–280. [Google Scholar] [CrossRef] [Green Version]
- Lancaster, M.A.; Knoblich, J.A. Organogenesis in a dish: Modeling development and disease using organoid technologies. Science 2014, 345, 1247125. [Google Scholar] [CrossRef] [PubMed]
- Subach, O.M.; Subach, F.V. GAF-CaMP3-sfGFP, An Enhanced Version of the Near-Infrared Genetically Encoded Positive Phytochrome-Based Calcium Indicator for the Visualization of Neuronal Activity. Int. J. Mol. Sci. 2020, 21, 6883. [Google Scholar] [CrossRef]
- Rossi, A.M.; Taylor, C.W. Reliable measurement of free Ca(2+) concentrations in the ER lumen using Mag-Fluo-4. Cell Calcium 2020, 87, 102188. [Google Scholar] [CrossRef] [PubMed]
- Helassa, N.; Durst, C.D.; Coates, C.; Kerruth, S.; Arif, U.; Schulze, C.; Wiegert, J.S.; Geeves, M.; Oertner, T.G.; Torok, K. Ultrafast glutamate sensors resolve high-frequency release at Schaffer collateral synapses. Proc. Natl. Acad. Sci. USA 2018, 115, 5594–5599. [Google Scholar] [CrossRef] [Green Version]
- Arai, S.; Kriszt, R.; Harada, K.; Looi, L.S.; Matsuda, S.; Wongso, D.; Suo, S.; Ishiura, S.; Tseng, Y.H.; Raghunath, M.; et al. RGB-Color Intensiometric Indicators to Visualize Spatiotemporal Dynamics of ATP in Single Cells. Angew. Chem. Int. Ed. Engl. 2018, 57, 10873–10878. [Google Scholar] [CrossRef]
- Barreto-Chang, O.L.; Dolmetsch, R.E. Calcium imaging of cortical neurons using Fura-2 AM. J. Vis. Exp. 2009, 23, e1067. [Google Scholar] [CrossRef]
- Richardson, D.S.; Gregor, C.; Winter, F.R.; Urban, N.T.; Sahl, S.J.; Willig, K.I.; Hell, S.W. SRpHi ratiometric pH biosensors for super-resolution microscopy. Nat. Commun. 2017, 8, 577. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.; Li, J.; Enterina, J.R.; Shen, Y.; Zhang, I.; Tewson, P.H.; Mo, G.C.; Zhang, J.; Quinn, A.M.; Hughes, T.E.; et al. Ratiometric biosensors based on dimerization-dependent fluorescent protein exchange. Nat. Methods 2015, 12, 195–198. [Google Scholar] [CrossRef]
- Cho, J.H.; Swanson, C.J.; Chen, J.; Li, A.; Lippert, L.G.; Boye, S.E.; Rose, K.; Sivaramakrishnan, S.; Chuong, C.M.; Chow, R.H. The GCaMP-R Family of Genetically Encoded Ratiometric Calcium Indicators. ACS Chem. Biol. 2017, 12, 1066–1074. [Google Scholar] [CrossRef] [Green Version]
- Paley, M.A.; Prescher, J.A. Bioluminescence: A versatile technique for imaging cellular and molecular features. MedChemComm 2014, 5, 255–267. [Google Scholar] [CrossRef] [Green Version]
- Yan, Y.; Shi, P.; Song, W.; Bi, S. Chemiluminescence and Bioluminescence Imaging for Biosensing and Therapy: In Vitro and In Vivo Perspectives. Theranostics 2019, 9, 4047–4065. [Google Scholar] [CrossRef]
- Alford, S.C.; Abdelfattah, A.S.; Ding, Y.; Campbell, R.E. A fluorogenic red fluorescent protein heterodimer. Chem. Biol. 2012, 19, 353–360. [Google Scholar] [CrossRef] [Green Version]
- Miller, K.E.; Kim, Y.; Huh, W.K.; Park, H.O. Bimolecular Fluorescence Complementation (BiFC) Analysis: Advances and Recent Applications for Genome-Wide Interaction Studies. J. Mol. Biol. 2015, 427, 2039–2055. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baird, G.S.; Zacharias, D.A.; Tsien, R.Y. Circular permutation and receptor insertion within green fluorescent proteins. Proc. Natl. Acad. Sci. USA 1999, 96, 11241–11246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Podor, B.; Hu, Y.L.; Ohkura, M.; Nakai, J.; Croll, R.; Fine, A. Comparison of genetically encoded calcium indicators for monitoring action potentials in mammalian brain by two-photon excitation fluorescence microscopy. Neurophotonics 2015, 2, 021014. [Google Scholar] [CrossRef] [Green Version]
- Shemesh, O.A.; Linghu, C.; Piatkevich, K.D.; Goodwin, D.; Celiker, O.T.; Gritton, H.J.; Romano, M.F.; Gao, R.; Yu, C.J.; Tseng, H.A.; et al. Precision Calcium Imaging of Dense Neural Populations via a Cell-Body-Targeted Calcium Indicator. Neuron 2020, 107, 470–486.e411. [Google Scholar] [CrossRef]
- Zou, Y.; Wang, A.; Huang, L.; Zhu, X.; Hu, Q.; Zhang, Y.; Chen, X.; Li, F.; Wang, Q.; Wang, H.; et al. Illuminating NAD(+) Metabolism in Live Cells and In Vivo Using a Genetically Encoded Fluorescent Sensor. Dev. Cell 2020, 53, 240–252. [Google Scholar] [CrossRef]
- Harada, K.; Ito, M.; Wang, X.; Tanaka, M.; Wongso, D.; Konno, A.; Hirai, H.; Hirase, H.; Tsuboi, T.; Kitaguchi, T. Red fluorescent protein-based cAMP indicator applicable to optogenetics and in vivo imaging. Sci. Rep. 2017, 7, 7351. [Google Scholar] [CrossRef] [Green Version]
- Kitajima, N.; Takikawa, K.; Sekiya, H.; Satoh, K.; Asanuma, D.; Sakamoto, H.; Takahashi, S.; Hanaoka, K.; Urano, Y.; Namiki, S.; et al. Real-time in vivo imaging of extracellular ATP in the brain with a hybrid-type fluorescent sensor. Elife 2020, 9, e57544. [Google Scholar] [CrossRef]
- Lobas, M.A.; Tao, R.; Nagai, J.; Kronschlager, M.T.; Borden, P.M.; Marvin, J.S.; Looger, L.L.; Khakh, B.S. A genetically encoded single-wavelength sensor for imaging cytosolic and cell surface ATP. Nat. Commun. 2019, 10, 711. [Google Scholar] [CrossRef] [Green Version]
- Marvin, J.S.; Scholl, B.; Wilson, D.E.; Podgorski, K.; Kazemipour, A.; Muller, J.A.; Schoch, S.; Quiroz, F.J.U.; Rebola, N.; Bao, H.; et al. Stability, affinity, and chromatic variants of the glutamate sensor iGluSnFR. Nat. Methods 2018, 15, 936–939. [Google Scholar] [CrossRef]
- Marvin, J.S.; Shimoda, Y.; Magloire, V.; Leite, M.; Kawashima, T.; Jensen, T.P.; Kolb, I.; Knott, E.L.; Novak, O.; Podgorski, K.; et al. A genetically encoded fluorescent sensor for in vivo imaging of GABA. Nat. Methods 2019, 16, 763–770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Terskikh, A.; Fradkov, A.; Ermakova, G.; Zaraisky, A.; Tan, P.; Kajava, A.V.; Zhao, X.; Lukyanov, S.; Matz, M.; Kim, S.; et al. “Fluorescent timer”: Protein that changes color with time. Science 2000, 290, 1585–1588. [Google Scholar] [CrossRef] [PubMed]
- Berlin, S.; Carroll, E.C.; Newman, Z.L.; Okada, H.O.; Quinn, C.M.; Kallman, B.; Rockwell, N.C.; Martin, S.S.; Lagarias, J.C.; Isacoff, E.Y. Photoactivatable genetically encoded calcium indicators for targeted neuronal imaging. Nat. Methods 2015, 12, 852–858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rainey, K.H.; Patterson, G.H. Photoswitching FRET to monitor protein-protein interactions. Proc. Natl. Acad. Sci. USA 2019, 116, 864–873. [Google Scholar] [CrossRef] [Green Version]
- Adam, V.; Berardozzi, R.; Byrdin, M.; Bourgeois, D. Phototransformable fluorescent proteins: Future challenges. Curr. Opin. Chem. Biol. 2014, 20, 92–102. [Google Scholar] [CrossRef]
- Zhang, X.; Hu, Y.; Yang, X.; Tang, Y.; Han, S.; Kang, A.; Deng, H.; Chi, Y.; Zhu, D.; Lu, Y. FOrster resonance energy transfer (FRET)-based biosensors for biological applications. Biosens. Bioelectron. 2019, 138, 111314. [Google Scholar] [CrossRef] [PubMed]
- Margineanu, A.; Chan, J.J.; Kelly, D.J.; Warren, S.C.; Flatters, D.; Kumar, S.; Katan, M.; Dunsby, C.W.; French, P.M. Screening for protein-protein interactions using Forster resonance energy transfer (FRET) and fluorescence lifetime imaging microscopy (FLIM). Sci. Rep. 2016, 6, 28186. [Google Scholar] [CrossRef] [Green Version]
- Salahpour, A.; Espinoza, S.; Masri, B.; Lam, V.; Barak, L.S.; Gainetdinov, R.R. BRET biosensors to study GPCR biology, pharmacology, and signal transduction. Front. Endocrinol. (Lausanne) 2012, 3, 105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De, A.; Jasani, A.; Arora, R.; Gambhir, S.S. Evolution of BRET Biosensors from Live Cell to Tissue-Scale In vivo Imaging. Front. Endocrinol. (Lausanne) 2013, 4, 131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, J.; Burgess, K. Fluorescent indicators for intracellular pH. Chem. Rev. 2010, 110, 2709–2728. [Google Scholar] [CrossRef]
- Nicholls, D.G. Fluorescence measurement of mitochondrial membrane potential changes in cultured cells. Methods Mol. Biol. 2012, 810, 119–133. [Google Scholar]
- Wiederschain, G.Y. The Molecular Probes handbook. A guide to fluorescent probes and labeling technologies. Biochemistry 2011, 76, 1276. [Google Scholar]
- Paredes, R.M.; Etzler, J.C.; Watts, L.T.; Zheng, W.; Lechleiter, J.D. Chemical calcium indicators. Methods 2008, 46, 143–151. [Google Scholar] [CrossRef] [Green Version]
- Palmer, A.E.; Tsien, R.Y. Measuring calcium signaling using genetically targetable fluorescent indicators. Nat. Protoc. 2006, 1, 1057–1065. [Google Scholar] [CrossRef]
- Shuai, J.; Parker, I. Optical single-channel recording by imaging Ca2+ flux through individual ion channels: Theoretical considerations and limits to resolution. Cell Calcium 2005, 37, 283–299. [Google Scholar] [CrossRef] [PubMed]
- Prilloff, S.; Noblejas, M.I.; Chedhomme, V.; Sabel, B.A. Two faces of calcium activation after optic nerve trauma: Life or death of retinal ganglion cells in vivo depends on calcium dynamics. Eur. J. Neurosci. 2007, 25, 3339–3346. [Google Scholar] [CrossRef] [PubMed]
- Galifianakis, N.V.; Placantonakis, D.G.; Chesler, M. Intracellular pH Measurements in Glioblastoma Cells Using the pH-Sensitive Dye BCECF. Methods Mol. Biol. 2018, 1741, 103–109. [Google Scholar] [PubMed]
- Ramshesh, V.K.; Lemasters, J.J. Imaging of Mitochondrial pH Using SNARF-1. Methods Mol. Biol. 2018, 1782, 351–356. [Google Scholar]
- Le Guern, F.; Mussard, V.; Gaucher, A.; Rottman, M.; Prim, D. Fluorescein Derivatives as Fluorescent Probes for pH Monitoring along Recent Biological Applications. Int. J. Mol. Sci. 2020, 21, 9217. [Google Scholar] [CrossRef] [PubMed]
- Lucien, F.; Harper, K.; Pelletier, P.P.; Volkov, L.; Dubois, C.M. Simultaneous pH measurement in endocytic and cytosolic compartments in living cells using confocal microscopy. J. Vis. Exp. 2014, 86, 51395. [Google Scholar] [CrossRef] [Green Version]
- Jin, L.; Millard, A.C.; Wuskell, J.P.; Clark, H.A.; Loew, L.M. Cholesterol-enriched lipid domains can be visualized by di-4-ANEPPDHQ with linear and nonlinear optics. Biophys. J. 2005, 89, L04–L06. [Google Scholar] [CrossRef] [Green Version]
- Owen, D.M.; Lanigan, P.M.; Dunsby, C.; Munro, I.; Grant, D.; Neil, M.A.; French, P.M.; Magee, A.I. Fluorescence lifetime imaging provides enhanced contrast when imaging the phase-sensitive dye di-4-ANEPPDHQ in model membranes and live cells. Biophys. J. 2006, 90, L80–L82. [Google Scholar] [CrossRef] [Green Version]
- Ronot, X.; Benel, L.; Adolphe, M.; Mounolou, J.C. Mitochondrial analysis in living cells: The use of rhodamine 123 and flow cytometry. Biol. Cell 1986, 57, 1–7. [Google Scholar] [CrossRef]
- Scaduto, R.C., Jr.; Grotyohann, L.W. Measurement of mitochondrial membrane potential using fluorescent rhodamine derivatives. Biophys. J. 1999, 76, 469–477. [Google Scholar] [CrossRef] [Green Version]
- Huang, S.G. Development of a high throughput screening assay for mitochondrial membrane potential in living cells. J. Biomol. Screen. 2002, 7, 383–389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyawaki, A.; Griesbeck, O.; Heim, R.; Tsien, R.Y. Dynamic and quantitative Ca2+ measurements using improved cameleons. Proc. Natl. Acad. Sci. USA 1999, 96, 2135–2140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cong, L.; Ran, F.A.; Cox, D.; Lin, S.; Barretto, R.; Habib, N.; Hsu, P.D.; Wu, X.; Jiang, W.; Marraffini, L.A.; et al. Multiplex genome engineering using CRISPR/Cas systems. Science 2013, 339, 819–823. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kotin, R.M.; Linden, R.M.; Berns, K.I. Characterization of a preferred site on human chromosome 19q for integration of adeno-associated virus DNA by non-homologous recombination. EMBO J. 1992, 11, 5071–5078. [Google Scholar] [CrossRef] [PubMed]
- Irion, S.; Luche, H.; Gadue, P.; Fehling, H.J.; Kennedy, M.; Keller, G. Identification and targeting of the ROSA26 locus in human embryonic stem cells. Nat. Biotechnol. 2007, 25, 1477–1482. [Google Scholar] [CrossRef]
- Liu, R.; Paxton, W.A.; Choe, S.; Ceradini, D.; Martin, S.R.; Horuk, R.; MacDonald, M.E.; Stuhlmann, H.; Koup, R.A.; Landau, N.R. Homozygous defect in HIV-1 coreceptor accounts for resistance of some multiply-exposed individuals to HIV-1 infection. Cell 1996, 86, 367–377. [Google Scholar] [CrossRef] [Green Version]
- Pellenz, S.; Phelps, M.; Tang, W.; Hovde, B.T.; Sinit, R.B.; Fu, W.; Li, H.; Chen, E.; Monnat, R.J., Jr. New Human Chromosomal Sites with "Safe Harbor" Potential for Targeted Transgene Insertion. Hum. Gene Ther. 2019, 30, 814–828. [Google Scholar] [CrossRef] [PubMed]
- Tiyaboonchai, A.; Mac, H.; Shamsedeen, R.; Mills, J.A.; Kishore, S.; French, D.L.; Gadue, P. Utilization of the AAVS1 safe harbor locus for hematopoietic specific transgene expression and gene knockdown in human ES cells. Stem Cell Res. 2014, 12, 630–637. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, Z.; Zhu, P.; Shi, H.; Guo, L.; Zhang, Q.; Chen, Y.; Chen, S.; Zhang, Z.; Peng, J.; Chen, J. PTC-bearing mRNA elicits a genetic compensation response via Upf3a and COMPASS components. Nature 2019, 568, 259–263. [Google Scholar] [CrossRef]
- El-Brolosy, M.A.; Kontarakis, Z.; Rossi, A.; Kuenne, C.; Gunther, S.; Fukuda, N.; Kikhi, K.; Boezio, G.L.M.; Takacs, C.M.; Lai, S.L.; et al. Genetic compensation triggered by mutant mRNA degradation. Nature 2019, 568, 193–197. [Google Scholar] [CrossRef]
- Kallunki, T.; Barisic, M.; Jaattela, M.; Liu, B. How to Choose the Right Inducible Gene Expression System for Mammalian Studies? Cells 2019, 8, 796. [Google Scholar] [CrossRef] [Green Version]
- Sutermaster, B.A.; Darling, E.M. Considerations for high-yield, high-throughput cell enrichment: Fluorescence versus magnetic sorting. Sci. Rep. 2019, 9, 227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Griffin, B.A.; Adams, S.R.; Tsien, R.Y. Specific covalent labeling of recombinant protein molecules inside live cells. Science 1998, 281, 269–272. [Google Scholar] [CrossRef] [Green Version]
- Xu, S.; Hu, H.Y. Fluorogen-activating proteins: Beyond classical fluorescent proteins. Acta Pharm. Sin. B 2018, 8, 339–348. [Google Scholar] [CrossRef]
- Gallo, E. Fluorogen-Activating Proteins: Next-Generation Fluorescence Probes for Biological Research. Bioconjug. Chem. 2020, 31, 16–27. [Google Scholar] [CrossRef] [PubMed]
- Liss, V.; Barlag, B.; Nietschke, M.; Hensel, M. Self-labelling enzymes as universal tags for fluorescence microscopy, super-resolution microscopy and electron microscopy. Sci. Rep. 2015, 5, 17740. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; So, M.K.; Loening, A.M.; Yao, H.; Gambhir, S.S.; Rao, J. HaloTag protein-mediated site-specific conjugation of bioluminescent proteins to quantum dots. Angew. Chem. Int. Ed. Engl. 2006, 45, 4936–4940. [Google Scholar] [CrossRef] [PubMed]
- Gautier, A.; Juillerat, A.; Heinis, C.; Correa, I.R., Jr.; Kindermann, M.; Beaufils, F.; Johnsson, K. An engineered protein tag for multiprotein labeling in living cells. Chem. Biol. 2008, 15, 128–136. [Google Scholar] [CrossRef] [Green Version]
- Pfleger, K.D.; Eidne, K.A. Illuminating insights into protein-protein interactions using bioluminescence resonance energy transfer (BRET). Nat. Methods 2006, 3, 165–174. [Google Scholar] [CrossRef]
- Baens, M.; Noels, H.; Broeckx, V.; Hagens, S.; Fevery, S.; Billiau, A.D.; Vankelecom, H.; Marynen, P. The dark side of EGFP: Defective polyubiquitination. PLoS ONE 2006, 1, e54. [Google Scholar] [CrossRef] [Green Version]
- Breckwoldt, M.O.; Pfister, F.M.; Bradley, P.M.; Marinkovic, P.; Williams, P.R.; Brill, M.S.; Plomer, B.; Schmalz, A.; St Clair, D.K.; Naumann, R.; et al. Multiparametric optical analysis of mitochondrial redox signals during neuronal physiology and pathology in vivo. Nat. Med. 2014, 20, 555–560. [Google Scholar] [CrossRef]
- Stapper, Z.A.; Jahn, T.R. Changes in Glutathione Redox Potential Are Linked to Abeta42-Induced Neurotoxicity. Cell Rep. 2018, 24, 1696–1703. [Google Scholar] [CrossRef] [Green Version]
- Lin, W.; Mehta, S.; Zhang, J. Genetically encoded fluorescent biosensors illuminate kinase signaling in cancer. J. Biol. Chem. 2019, 294, 14814–14822. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stoeber, M.; Jullie, D.; Lobingier, B.T.; Laeremans, T.; Steyaert, J.; Schiller, P.W.; Manglik, A.; von Zastrow, M. A Genetically Encoded Biosensor Reveals Location Bias of Opioid Drug Action. Neuron 2018, 98, 963–976. [Google Scholar] [CrossRef] [Green Version]
- Pak, V.V.; Ezerina, D.; Lyublinskaya, O.G.; Pedre, B.; Tyurin-Kuzmin, P.A.; Mishina, N.M.; Thauvin, M.; Young, D.; Wahni, K.; Martinez Gache, S.A.; et al. Ultrasensitive Genetically Encoded Indicator for Hydrogen Peroxide Identifies Roles for the Oxidant in Cell Migration and Mitochondrial Function. Cell Metab. 2020, 31, 642–653. [Google Scholar] [CrossRef]
- Dong, C.; Ly, C.; Dunlap, L.E.; Vargas, M.V.; Sun, J.; Hwang, I.W.; Azinfar, A.; Oh, W.C.; Wetsel, W.C.; Olson, D.E.; et al. Psychedelic-inspired drug discovery using an engineered biosensor. Cell 2021, 184, 2779–2792. [Google Scholar] [CrossRef] [PubMed]
- Morris, M.C. Fluorescent biosensors—probing protein kinase function in cancer and drug discovery. Biochim. Biophys. Acta 2013, 1834, 1387–1395. [Google Scholar] [CrossRef]
- Misra, T.; Baccino-Calace, M.; Meyenhofer, F.; Rodriguez-Crespo, D.; Akarsu, H.; Armenta-Calderon, R.; Gorr, T.A.; Frei, C.; Cantera, R.; Egger, B.; et al. A genetically encoded biosensor for visualising hypoxia responses in vivo. Biol. Open 2017, 6, 296–304. [Google Scholar] [PubMed] [Green Version]
- Shivange, A.V.; Borden, P.M.; Muthusamy, A.K.; Nichols, A.L.; Bera, K.; Bao, H.; Bishara, I.; Jeon, J.; Mulcahy, M.J.; Cohen, B.; et al. Determining the pharmacokinetics of nicotinic drugs in the endoplasmic reticulum using biosensors. J. Gen. Physiol. 2019, 151, 738–757. [Google Scholar] [CrossRef] [PubMed]
- Komatsu, N.; Terai, K.; Imanishi, A.; Kamioka, Y.; Sumiyama, K.; Jin, T.; Okada, Y.; Nagai, T.; Matsuda, M. A platform of BRET-FRET hybrid biosensors for optogenetics, chemical screening, and in vivo imaging. Sci. Rep. 2018, 8, 8984. [Google Scholar] [CrossRef]
- Wehr, M.C.; Rossner, M.J. Split protein biosensor assays in molecular pharmacological studies. Drug Discov. Today 2016, 21, 415–429. [Google Scholar] [CrossRef] [PubMed]
- Berezin, M.Y.; Achilefu, S. Fluorescence lifetime measurements and biological imaging. Chem. Rev. 2010, 110, 2641–2684. [Google Scholar] [CrossRef] [PubMed] [Green Version]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Valetdinova, K.R.; Malankhanova, T.B.; Zakian, S.M.; Medvedev, S.P. The Cutting Edge of Disease Modeling: Synergy of Induced Pluripotent Stem Cell Technology and Genetically Encoded Biosensors. Biomedicines 2021, 9, 960. https://doi.org/10.3390/biomedicines9080960
Valetdinova KR, Malankhanova TB, Zakian SM, Medvedev SP. The Cutting Edge of Disease Modeling: Synergy of Induced Pluripotent Stem Cell Technology and Genetically Encoded Biosensors. Biomedicines. 2021; 9(8):960. https://doi.org/10.3390/biomedicines9080960
Chicago/Turabian StyleValetdinova, Kamila R., Tuyana B. Malankhanova, Suren M. Zakian, and Sergey P. Medvedev. 2021. "The Cutting Edge of Disease Modeling: Synergy of Induced Pluripotent Stem Cell Technology and Genetically Encoded Biosensors" Biomedicines 9, no. 8: 960. https://doi.org/10.3390/biomedicines9080960