
In Vitro Nephrotoxicity Studies of Established and Experimental Platinum-Based Compounds


Abstract
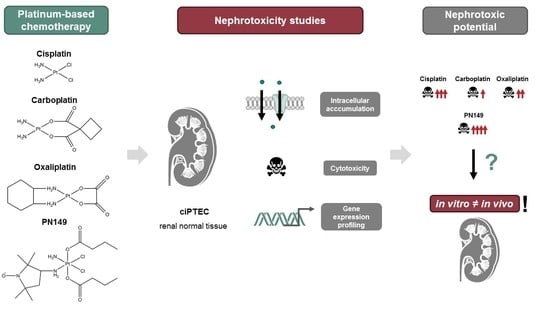
:
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Platinum-Based Compounds
2.3. Cytotoxicity Studies
2.4. Intracellular Platinum Accumulation
2.5. Gene Expression Profiling by High-Throughput RT-qPCR
2.6. Spectrophotometric Measurement of Anti-Oxidant Capacity of Platinum Compounds
3. Results and Discussion
3.1. Experimental Models for In Vitro Nephrotoxicity Studies
3.2. Cytotoxic Potential of Platinum Compounds in ciPTEC
3.3. Intracellular Platinum Accumulation
3.4. Gene Expression Profiling
3.4.1. Impact of Platinum Complexes on Genes Coding for Transcription Factors
3.4.2. Impact of Platinum Complexes on Genes Related to Oxidative Stress Response and Inflammation
3.4.3. Impact of Platinum Complexes on Genes Related to Cell Cycle Control
3.4.4. Impact of Platinum Complexes on Genes Related to DNA Damage Response/Repair and Apoptosis
3.5. Comparison of Gene Expression Profiles of Cisplatin in ciPTECs vs. Normal Renal Tissue Cells
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AAPH | 2,2′-azobis(2-amidinopropane) dihydrochloride |
| AAS | atomic absorption spectroscopy |
| AKI | acute kidney injury |
| DDR | DNA damage response |
| DSB | double strand break |
| ICL | interstrand crosslink |
| PB | phosphate buffer |
| PNC | platinum(IV)–nitroxyl complex |
| PN149 | e-ammine-d-(3-amino-2,2,5,5-tetramethylpyrrolidine-1-oxyl)-a,f-bis(butanoato)-b,c-dichloroplatinum(IV) |
| PTECs | proximal tubule epithelial cells |
| RCC | relative cell count |
| ROS | reactive oxygen species |
| SLC | solute carrier |
References
- Dilruba, S.; Kalayda, G.V. Platinum-based drugs: Past, present and future. Cancer Chemother. Pharmacol. 2016, 77, 1103–1124. [Google Scholar] [CrossRef]
- Masters, J.R.; Köberle, B. Curing metastatic cancer: Lessons from testicular germ-cell tumours. Nat. Rev. Cancer 2003, 3, 517–525. [Google Scholar] [CrossRef]
- Oun, R.; Moussa, Y.E.; Wheate, N.J. The side effects of platinum-based chemotherapy drugs: A review for chemists. Dalton Trans. 2018, 47, 6645–6653. [Google Scholar] [CrossRef]
- Manohar, S.; Leung, N. Cisplatin nephrotoxicity: A review of the literature. J. Nephrol. 2017, 31, 15–25. [Google Scholar] [CrossRef]
- Basu, A.; Krishnamurthy, S. Cellular Responses to Cisplatin-Induced DNA Damage. J. Nucleic Acids 2010, 2010, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Siddik, Z.H. Cisplatin: Mode of cytotoxic action and molecular basis of resistance. Oncogene 2003, 22, 7265–7279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Florea, A.-M.; Büsselberg, D. Cisplatin as an Anti-Tumor Drug: Cellular Mechanisms of Activity, Drug Resistance and Induced Side Effects. Cancers 2011, 3, 1351–1371. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Lippard, S.J. Cellular processing of platinum anticancer drugs. Nat. Rev. Drug Discov. 2005, 4, 307–320. [Google Scholar] [CrossRef] [PubMed]
- Hall, M.D.; Okabe, M.; Shen, D.-W.; Liang, X.-J.; Gottesman, M.M. The Role of Cellular Accumulation in Determining Sensitivity to Platinum-Based Chemotherapy. Annu. Rev. Pharmacol. Toxicol. 2008, 48, 495–535. [Google Scholar] [CrossRef]
- Spreckelmeyer, S.; Orvig, C.; Casini, A. Cellular Transport Mechanisms of Cytotoxic Metallodrugs: An Overview beyond Cisplatin. Molecules 2014, 19, 15584–15610. [Google Scholar] [CrossRef] [Green Version]
- Kelland, L. The resurgence of platinum-based cancer chemotherapy. Nat. Rev. Cancer 2007, 7, 573–584. [Google Scholar] [CrossRef]
- Dugbartey, G.J.; Peppone, L.J.; de Graaf, I.A. An integrative view of cisplatin-induced renal and cardiac toxicities: Molecular mechanisms, current treatment challenges and potential protective measures. Toxicology 2016, 371, 58–66. [Google Scholar] [CrossRef] [Green Version]
- Pabla, N.; Dong, Z. Cisplatin nephrotoxicity: Mechanisms and renoprotective strategies. Kidney Int. 2008, 73, 994–1007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yao, X.; Panichpisal, K.; Kurtzman, N.; Nugent, K. Cisplatin Nephrotoxicity: A Review. Am. J. Med Sci. 2007, 334, 115–124. [Google Scholar] [CrossRef] [Green Version]
- Aydinoz, S.; Uzun, G.; Cermik, H.; Atasoyu, E.M.; Yildiz, S.; Karagoz, B.; Evrenkaya, R. Effects of Different Doses of Hyperbaric Oxygen on Cisplatin-Induced Nephrotoxicity. Ren. Fail. 2007, 29, 257–263. [Google Scholar] [CrossRef]
- Deng, J.; Kohda, Y.; Chiao, H.; Wang, Y.; Hu, X.; Hewitt, S.; Miyaji, T.; Mcleroy, P.; Nibhanupudy, B.; Li, S.; et al. Interleukin-10 inhibits ischemic and cisplatin-induced acute renal injury. Kidney Int. 2001, 60, 2118–2128. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Hanigan, M.H. Role of CysteineS-Conjugate β-Lyase in the Metabolism of Cisplatin. J. Pharmacol. Exp. Ther. 2003, 306, 988–994. [Google Scholar] [CrossRef] [Green Version]
- Townsend, D.M.; Deng, M.; Zhang, L.; Lapus, M.G.; Hanigan, M.H. Metabolism of Cisplatin to a Nephrotoxin in Proximal Tubule Cells. J. Am. Soc. Nephrol. 2003, 14, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wainford, R.D.; Weaver, R.J.; Stewart, K.N.; Brown, P.; Hawksworth, G.M. Cisplatin nephrotoxicity is mediated by gamma glutamyltranspeptidase, not via a C-S lyase governed biotransformation pathway. Toxicology 2008, 249, 184–193. [Google Scholar] [CrossRef] [PubMed]
- Johnstone, T.; Suntharalingam, K.; Lippard, S.J. The Next Generation of Platinum Drugs: Targeted Pt(II) Agents, Nanoparticle Delivery, and Pt(IV) Prodrugs. Chem. Rev. 2016, 116, 3436–3486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barnard, C. Platinum Group Metal Compounds in Cancer Chemotherapy. Johns. Matthey Technol. Rev. 2017, 61, 52–59. [Google Scholar] [CrossRef]
- Köberle, B.; Schoch, S. Platinum Complexes in Colorectal Cancer and Other Solid Tumors. Cancers 2021, 13, 2073. [Google Scholar] [CrossRef] [PubMed]
- Wheate, N.J.; Walker, S.; Craig, G.E.; Oun, R. The status of platinum anticancer drugs in the clinic and in clinical trials. Dalton Trans. 2010, 39, 8113–8127. [Google Scholar] [CrossRef] [Green Version]
- Kelland, L. Broadening the clinical use of platinum drug–based chemotherapy with new analogues. Expert Opin. Investig. Drugs 2007, 16, 1009–1021. [Google Scholar] [CrossRef]
- Sen, V.D.; Tkachev, V.V.; Volkova, L.M.; Goncharova, S.A.; Raevskaya, T.A.; Konovalova, N.P. Synthesis, structure, and antitumor properties of platinum(iv) complexes with aminonitroxyl radicals. Russ. Chem. Bull. 2003, 52, 421–426. [Google Scholar] [CrossRef]
- Sen, V.D.; Rukina, N.A.; Tkachev, V.V.; PisMenskii, A.V.; Volkova, L.M.; Goncharova, S.A.; Raevskaya, T.A.; Tikhomirov, A.G.; Gorbacheva, L.B.; Konovalova, N.P. Synthesis, structure, and biological activity of mixed-ligand platinum(II) complexes with aminonitroxides. Russ. Chem. Bull. 2000, 49, 1613–1619. [Google Scholar] [CrossRef]
- Sen, V.D.; Golubev, V.A.; Lugovskaya, N.Y.; Sashenkova, T.E.; Konovalova, N.P. Synthesis and antitumor properties of new platinum(IV) complexes with aminonitroxyl radicals. Russ. Chem. Bull. 2006, 55, 62–65. [Google Scholar] [CrossRef]
- Cetraz, M.; Sen, V.; Schoch, S.; Streule, K.; Golubev, V.; Hartwig, A.; Köberle, B. Platinum(IV)-nitroxyl complexes as possible candidates to circumvent cisplatin resistance in RT112 bladder cancer cells. Arch. Toxicol. 2016, 91, 785–797. [Google Scholar] [CrossRef]
- Schoch, S.; Sen, V.; Gajewski, S.; Golubev, V.; Strauch, B.; Hartwig, A.; Köberle, B. Activity profile of the cisplatin analogue PN149 in different tumor cell lines. Biochem. Pharmacol. 2018, 156, 109–119. [Google Scholar] [CrossRef] [PubMed]
- Fischer, B.M.; Neumann, D.; Piberger, A.L.; Risnes, S.F.; Köberle, B.; Hartwig, A. Use of high-throughput RT-qPCR to assess modulations of gene expression profiles related to genomic stability and interactions by cadmium. Arch. Toxicol. 2015, 90, 2745–2761. [Google Scholar] [CrossRef] [Green Version]
- Wilmer, M.J.; Saleem, M.A.; Masereeuw, R.; Ni, L.; Van Der Velden, T.J.; Russel, F.; Mathieson, P.W.; Monnens, L.A.; Heuvel, L.P.V.D.; Levtchenko, E.N. Novel conditionally immortalized human proximal tubule cell line expressing functional influx and efflux transporters. Cell Tissue Res. 2009, 339, 449–457. [Google Scholar] [CrossRef] [Green Version]
- O’Dwyer, P.J.; Stevenson, J.P.; Johnson, S.W. Clinical Pharmacokinetics and Administration of Established Platinum Drugs. Drugs 2000, 59, 19–27. [Google Scholar] [CrossRef] [PubMed]
- Schierl, R.; Rohrer, B. Long-term platinum excretion in patients treated with cisplatin. Cancer Chemother. Pharmacol. 1995, 36, 75–78. [Google Scholar] [CrossRef]
- De Jongh, F.E.; Gallo, J.M.; Shen, M.; Verweij, J.; Sparreboom, A. Population pharmacokinetics of cisplatin in adult cancer patients. Cancer Chemother. Pharmacol. 2004, 54, 105–112. [Google Scholar] [CrossRef] [PubMed]
- Takashima, M.; Horie, M.; Shichiri, M.; Hagihara, Y.; Yoshida, Y.; Niki, E. Assessment of antioxidant capacity for scavenging free radicals in vitro: A rational basis and practical application. Free. Radic. Biol. Med. 2012, 52, 1242–1252. [Google Scholar] [CrossRef] [PubMed]
- Giandomenico, C.M.; Abrams, M.J.; Murrer, B.A.; Vollano, J.F.; Rheinheimer, M.I.; Wyer, S.B.; Bossard, G.E.; Higgins, J.D. Carboxylation of Kinetically Inert Platinum(IV) Hydroxy Complexes. An Entr.acte.ee into Orally Active Platinum(IV) Antitumor Agents. Inorg. Chem. 1995, 34, 1015–1021. [Google Scholar] [CrossRef] [PubMed]
- Jang, K.-J.; Mehr, A.P.; Hamilton, G.A.; McPartlin, L.A.; Chung, S.; Suh, K.-Y.; Ingber, D.E. Human kidney proximal tubule-on-a-chip for drug transport and nephrotoxicity assessment. Integr. Biol. 2013, 5, 1119–1129. [Google Scholar] [CrossRef]
- Tiong, H.Y.; Huang, P.; Xiong, S.; Li, Y.; Vathsala, A.; Zink, D. Drug-Induced Nephrotoxicity: Clinical Impact and Preclinicalin VitroModels. Mol. Pharm. 2014, 11, 1933–1948. [Google Scholar] [CrossRef]
- Sánchez-Romero, N.; Schophuizen, C.M.; Giménez, I.; Masereeuw, R. In vitro systems to study nephropharmacology: 2D versus 3D models. Eur. J. Pharmacol. 2016, 790, 36–45. [Google Scholar] [CrossRef]
- Shaw, G.; Morse, S.; Ararat, M.; Graham, F.L. Preferential transformation of human neuronal cells by human adenoviruses and the origin of HEK 293 cells. FASEB J. 2002, 16, 869–871. [Google Scholar] [CrossRef]
- Jenkinson, S.E.; Chung, G.W.; Van Loon, E.; Bakar, N.S.; Dalzell, A.M.; Brown, C.D.A. The limitations of renal epithelial cell line HK-2 as a model of drug transporter expression and function in the proximal tubule. Pflug. Arch. 2012, 464, 601–611. [Google Scholar] [CrossRef]
- Filipski, K.K.; Loos, W.J.; Verweij, J.; Sparreboom, A. Interaction of Cisplatin with the Human Organic Cation Transporter 2. Clin. Cancer Res. 2008, 14, 3875–3880. [Google Scholar] [CrossRef] [Green Version]
- Zu Schwabedissen, H.E.M.; Verstuyft, C.; Kroemer, H.K.; Becquemont, L.; Kim, R.B. Human multidrug and toxin extrusion 1 (MATE1/SLC47A1) transporter: Functional characterization, interaction with OCT2 (SLC22A2), and single nucleotide polymorphisms. Am. J. Physiol. Physiol. 2010, 298, F997–F1005. [Google Scholar] [CrossRef] [Green Version]
- Wieser, M.; Stadler, G.; Jennings, P.; Streubel, B.; Pfaller, W.; Ambros, P.; Riedl, C.; Katinger, H.; Grillari, R.; Grillari-Voglauer, R. hTERT alone immortalizes epithelial cells of renal proximal tubules without changing their functional characteristics. Am. J. Physiol. Physiol. 2008, 295, F1365–F1375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nieskens, T.T.G.; Peters, J.G.P.; Schreurs, M.J.; Smits, N.; Woestenenk, R.; Jansen, K.; van der Made, T.K.; Röring, M.; Hilgendorf, C.; Wilmer, M.J.; et al. A Human Renal Proximal Tubule Cell Line with Stable Organic Anion Transporter 1 and 3 Expression Predictive for Antiviral-Induced Toxicity. AAPS J. 2016, 18, 465–475. [Google Scholar] [CrossRef] [Green Version]
- Secker, P. RPTEC/TERT1 cells form highly differentiated tubules when cultured in a 3D matrix. ALTEX 2018, 35, 223–234. [Google Scholar] [CrossRef]
- Zhang, S.; Lovejoy, K.S.; Shima, J.E.; Lagpacan, L.L.; Shu, Y.; Lapuk, A.; Chen, Y.; Komori, T.; Gray, J.W.; Chen, X.; et al. Organic Cation Transporters Are Determinants of Oxaliplatin Cytotoxicity. Cancer Res. 2006, 66, 8847–8857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krüger, K.; Thomale, J.; Stojanović, N.; Osmak, M.; Henninger, C.; Bormann, S.; Fritz, G. Platinum-induced kidney damage: Unraveling the DNA damage response (DDR) of renal tubular epithelial and glomerular endothelial cells following platinum injury. Biochim. Biophys. Acta 2015, 1853, 685–698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yokoo, S.; Yonezawa, A.; Masuda, S.; Fukatsu, A.; Katsura, T.; Inui, K.-I. Differential contribution of organic cation transporters, OCT2 and MATE1, in platinum agent-induced nephrotoxicity. Biochem. Pharmacol. 2007, 74, 477–487. [Google Scholar] [CrossRef]
- Safirstein, R.; Miller, P.; Guttenplan, J. Uptake and metabolism of cisplatin by rat kidney. Kidney Int. 1984, 25, 753–758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- dos Santos, N.A.G.; Rodrigues, M.A.C.; Martins, N.M.; Santos, A.C. Cisplatin-induced nephrotoxicity and targets of nephroprotection: An update. Arch. Toxicol. 2012, 86, 1233–1250. [Google Scholar] [CrossRef]
- Staud, F.; Cerveny, L.; Ahmadimoghaddam, D.; Ceckova, M. Multidrug and toxin extrusion proteins (MATE/SLC47); role in pharmacokinetics. Int. J. Biochem. Cell Biol. 2013, 45, 2007–2011. [Google Scholar] [CrossRef] [PubMed]
- Ciarimboli, G.; Ludwig, T.; Lang, D.; Pavenstädt, H.; Koepsell, H.; Piechota, H.-J.; Haier, J.; Jaehde, U.; Zisowsky, J.; Schlatter, E. Cisplatin Nephrotoxicity Is Critically Mediated via the Human Organic Cation Transporter 2. Am. J. Pathol. 2005, 167, 1477–1484. [Google Scholar] [CrossRef] [Green Version]
- Pabla, N.; Murphy, R.F.; Liu, K.; Dong, Z. The copper transporter Ctr1 contributes to cisplatin uptake by renal tubular cells during cisplatin nephrotoxicity. Am. J. Physiol. Physiol. 2009, 296, F505–F511. [Google Scholar] [CrossRef]
- Yonezawa, A.; Inui, K.-I. Organic cation transporter OCT/SLC22A and H+/organic cation antiporter MATE/SLC47A are key molecules for nephrotoxicity of platinum agents. Biochem. Pharmacol. 2011, 81, 563–568. [Google Scholar] [CrossRef]
- Yonezawa, A.; Masuda, S.; Yokoo, S.; Katsura, T.; Inui, K.-I. Cisplatin and Oxaliplatin, but Not Carboplatin and Nedaplatin, Are Substrates for Human Organic Cation Transporters (SLC22A1–3 and Multidrug and Toxin Extrusion Family). J. Pharmacol. Exp. Ther. 2006, 319, 879–886. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schoch, S.; Gajewski, S.; Rothfuß, J.; Hartwig, A.; Köberle, B. Comparative Study of the Mode of Action of Clinically Approved Platinum-Based Chemotherapeutics. Int. J. Mol. Sci. 2020, 21, 6928. [Google Scholar] [CrossRef] [PubMed]
- Ludwig, T.; Riethmüller, C.; Gekle, M.; Schwerdt, G.; Oberleithner, H. Nephrotoxicity of platinum complexes is related to basolateral organic cation transport. Kidney Int. 2004, 66, 196–202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aschauer, L.; Carta, G.; Vogelsang, N.; Schlatter, E.; Jennings, P. Expression of xenobiotic transporters in the human renal proximal tubule cell line RPTEC/TERT1. Toxicol. Vitr. 2015, 30, 95–105. [Google Scholar] [CrossRef]
- Secker, P.F.; Schlichenmaier, N.; Beilmann, M.; Deschl, U.; Dietrich, D.R. Functional transepithelial transport measurements to detect nephrotoxicity in vitro using the RPTEC/TERT1 cell line. Arch. Toxicol. 2019, 93, 1965–1978. [Google Scholar] [CrossRef]
- Sahni, V.; Choudhury, D.; Ahmed, Z. Chemotherapy-associated renal dysfunction. Nat. Rev. Nephrol. 2009, 5, 450–462. [Google Scholar] [CrossRef]
- Wilmes, A.; Bielow, C.; Ranninger, C.; Bellwon, P.; Aschauer, L.; Limonciel, A.; Chassaigne, H.; Kristl, T.; Aiche, S.; Huber, C.G.; et al. Mechanism of cisplatin proximal tubule toxicity revealed by integrating transcriptomics, proteomics, metabolomics and biokinetics. Toxicol. Vitr. 2015, 30, 117–127. [Google Scholar] [CrossRef] [PubMed]
- Shaulian, E.; Karin, M. AP-1 in cell proliferation and survival. Oncogene 2001, 20, 2390–2400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kobayashi, M.; Yamamoto, M. Molecular Mechanisms Activating the Nrf2-Keap1 Pathway of Antioxidant Gene Regulation. Antioxidants Redox Signal. 2005, 7, 385–394. [Google Scholar] [CrossRef] [PubMed]
- Chang, L.; Karin, M. Mammalian MAP kinase signalling cascades. Nature 2001, 410, 37–40. [Google Scholar] [CrossRef] [PubMed]
- Savinova, O.V.; Hoffmann, A.; Ghosh, G. The Nfkb1 and Nfkb2 Proteins p105 and p100 Function as the Core of High-Molecular-Weight Heterogeneous Complexes. Mol. Cell 2009, 34, 591–602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vyas, D.; Laput, G.; Vyas, A. Chemotherapy-enhanced inflammation may lead to the failure of therapy and metastasis. OncoTargets Ther. 2014, 7, 1015–1023. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shiraishi, F.; Curtis, L.M.; Truong, L.; Poss, K.; Visner, G.A.; Madsen, K.; Nick, H.S.; Agarwal, A. Heme oxygenase-1 gene ablation or expression modulates cisplatin-induced renal tubular apoptosis. Am. J. Physiol. Physiol. 2000, 278, F726–F736. [Google Scholar] [CrossRef] [Green Version]
- Durak, I.; Ozbek, H.; Karaayvaz, M.; Ozturk, H.S. Cisplatin induces acute renal failure by impairing antioxidant system in guinea pigs: Effects of antioxidant supplementation on the cisplatin nephrotoxicity. Drug Chem. Toxicol. 2002, 25, 1–8. [Google Scholar] [CrossRef]
- Tanabe, K.; Tamura, Y.; Lanaspa, M.A.; Miyazaki, M.; Suzuki, N.; Sato, W.; Maeshima, Y.; Schreiner, G.F.; Villarreal, F.J.; Johnson, R.J.; et al. Epicatechin limits renal injury by mitochondrial protection in cisplatin nephropathy. Am. J. Physiol. Physiol. 2012, 303, F1264–F1274. [Google Scholar] [CrossRef] [Green Version]
- Baek, S.M.; Kwon, C.H.; Kim, J.H.; Woo, J.S.; Jung, J.S.; Kim, Y.K. Differential roles of hydrogen peroxide and hydroxyl radical in cisplatin-induced cell death in renal proximal tubular epithelial cells. J. Lab. Clin. Med. 2003, 142, 178–186. [Google Scholar] [CrossRef]
- Tsutsumishita, Y.; Onda, T.; Okada, K.; Takeda, M.; Endou, H.; Futaki, S.; Niwa, M. Involvement of H2O2Production in Cisplatin-Induced Nephrotoxicity. Biochem. Biophys. Res. Commun. 1998, 242, 310–312. [Google Scholar] [CrossRef]
- Chirino, Y.I.; Pedraza-Chaverri, J. Role of oxidative and nitrosative stress in cisplatin-induced nephrotoxicity. Exp. Toxicol. Pathol. 2009, 61, 223–242. [Google Scholar] [CrossRef]
- Sen, V.D.; Terentiev, A.A.; Konovalova, N.P. Platinum Complexes with Bioactive Nitroxyl Radicals: Synthesis and Antitumor properties. In Nitroxides—Theory, Experiment and Applications; Kokorin, A., Ed.; InTech: Rijeka, Croatia, 2012. [Google Scholar] [CrossRef] [Green Version]
- Wilcox, C.S. Effects of tempol and redox-cycling nitroxides in models of oxidative stress. Pharmacol. Ther. 2010, 126, 119–145. [Google Scholar] [CrossRef] [Green Version]
- Lugrin, J.; Rosenblatt, N.; Parapanov, R.; Liaudet, L. The role of oxidative stress during inflammatory processes. Biol. Chem. 2014, 395, 203–230. [Google Scholar] [CrossRef] [Green Version]
- Schmitt, R.; Cantley, L.G. The impact of aging on kidney repair. Am. J. Physiol. Physiol. 2008, 294, F1265–F1272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Humphreys, B.D.; Czerniak, S.; DiRocco, D.P.; Hasnain, W.; Cheema, R.; Bonventre, J.V. Repair of injured proximal tubule does not involve specialized progenitors. Proc. Natl. Acad. Sci. USA 2011, 108, 9226–9231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, H.-Z.; Tsai, S.-Y.; Leone, G. Emerging roles of E2Fs in cancer: An exit from cell cycle control. Nat. Rev. Cancer 2009, 9, 785–797. [Google Scholar] [CrossRef] [Green Version]
- Baldin, V.; Lukas, J.; Marcote, M.J.; Pagano, M.; Draetta, G. Cyclin D1 is a nuclear protein required for cell cycle progression in G1. Genes Dev. 1993, 7, 812–821. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levens, D. Disentangling the MYC web. Proc. Natl. Acad. Sci. USA 2002, 99, 5757–5759. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karimian, A.; Ahmadi, Y.; Yousefi, B. Multiple functions of p21 in cell cycle, apoptosis and transcriptional regulation after DNA damage. DNA Repair 2016, 42, 63–71. [Google Scholar] [CrossRef]
- Ferenbach, D.; Bonventre, J.V. Mechanisms of maladaptive repair after AKI leading to accelerated kidney ageing and CKD. Nat. Rev. Nephrol. 2015, 11, 264–276. [Google Scholar] [CrossRef] [Green Version]
- Hanigan, M.H.; Devarajan, P. Cisplatin nephrotoxicity: Molecular mechanisms. Cancer Ther. 2003, 1, 47–61. [Google Scholar]
- Miyaji, T.; Kato, A.; Yasuda, H.; Fujigaki, Y.; Hishida, A. Role of the Increase in p21 in Cisplatin-Induced Acute Renal Failure in Rats. J. Am. Soc. Nephrol. 2001, 12, 900–908. [Google Scholar] [CrossRef] [PubMed]
- Huang, Q.; Dunn, R.T.; Jayadev, S.; DiSorbo, O.; Pack, F.D.; Farr, S.B.; Stoll, R.E.; Blanchard, K.T. Assessment of cisplatin-induced nephrotoxicity by microarray technology. Toxicol. Sci. 2001, 63, 196–207. [Google Scholar] [CrossRef] [Green Version]
- De Silva, I.U. Defects in interstrand cross-link uncoupling do not account for the extreme sensitivity of ERCC1 and XPF cells to cisplatin. Nucleic Acids Res. 2002, 30, 3848–3856. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Usanova, S.; Piée-Staffa, A.; Sied, U.; Thomale, J.; Schneider, A.; Kaina, B.; Köberle, B. Cisplatin sensitivity of testis tumour cells is due to deficiency in interstrand-crosslink repair and low ERCC1-XPF expression. Mol. Cancer 2010, 9, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clauson, C.; Schärer, O.D.; Niedernhofer, L. Advances in Understanding the Complex Mechanisms of DNA Interstrand Cross-Link Repair. Cold Spring Harb. Perspect. Biol. 2013, 5, a012732. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Keijzers, G.; Rasmussen, L.J. DNA mismatch repair and its many roles in eukaryotic cells. Mutat. Res. Mutat. Res. 2017, 773, 174–187. [Google Scholar] [CrossRef]
- Sobol, R.; Wilson, S. Mammalian DNA beta-polymerase in base excision repair of alkylation damage. Prog. Nucleic Acid Res. Mol. Biol. 2001, 68, 57–74. [Google Scholar] [CrossRef] [PubMed]
- Schärer, O.D. Nucleotide Excision Repair in Eukaryotes. Cold Spring Harb. Perspect. Biol. 2013, 5, a012609. [Google Scholar] [CrossRef]
- Ramesh, G.; Reeves, W. TNFR2-mediated apoptosis and necrosis in cisplatin-induced acute renal failure. Am. J. Physiol. Physiol. 2003, 285, F610–F618. [Google Scholar] [CrossRef]
- Pabla, N.; Huang, S.; Mi, Q.-S.; Daniel, R.; Dong, Z. ATR-Chk2 Signaling in p53 Activation and DNA Damage Response during Cisplatin-induced Apoptosis. J. Biol. Chem. 2008, 283, 6572–6583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fischer, M. Census and evaluation of p53 target genes. Oncogene 2017, 36, 3943–3956. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, M.; Wei, Q.; Wang, J.; Du, Q.; Yu, J.; Zhang, L.; Dong, Z. Regulation of PUMA-α by p53 in cisplatin-induced renal cell apoptosis. Oncogene 2006, 25, 4056–4066. [Google Scholar] [CrossRef] [Green Version]
- Nouspikel, T. DNA repair in terminally differentiated cells. DNA Repair 2002, 1, 59–75. [Google Scholar] [CrossRef]
- Benigni, A.; Morigi, M.; Remuzzi, G. Kidney regeneration. Lancet 2010, 375, 1310–1317. [Google Scholar] [CrossRef] [Green Version]
- Wilmer, M.J.; Ng, C.P.; Lanz, H.L.; Vulto, P.; Suter-Dick, L.; Masereeuw, R. Kidney-on-a-Chip Technology for Drug-Induced Nephrotoxicity Screening. Trends Biotechnol. 2015, 34, 156–170. [Google Scholar] [CrossRef]
- Chandrasekaran, V.; Carta, G.; Pereira, D.D.C.; Gupta, R.; Murphy, C.; Feifel, E.; Kern, G.; Lechner, J.; Cavallo, A.L.; Gupta, S.; et al. Generation and characterization of iPSC-derived renal proximal tubule-like cells with extended stability. Sci. Rep. 2021, 11, 1–17. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Platinum Compound | IC50 [µM] | ng Pt/106 Cells |
|---|---|---|
| Cisplatin | 13 | 17 ± 5 |
| PN149 Oxaliplatin Carboplatin | 6 51 175 | 244 ± 49 11 ± 2 18 ± 2 |
| Cellular Model | ng Pt/106 Cells |
|---|---|
| ciPTEC | 17 ± 5 |
| Primary kidney cells | 25 ± 5 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schoch, S.; Sen, V.; Brenner, W.; Hartwig, A.; Köberle, B. In Vitro Nephrotoxicity Studies of Established and Experimental Platinum-Based Compounds. Biomedicines 2021, 9, 1033. https://doi.org/10.3390/biomedicines9081033
Schoch S, Sen V, Brenner W, Hartwig A, Köberle B. In Vitro Nephrotoxicity Studies of Established and Experimental Platinum-Based Compounds. Biomedicines. 2021; 9(8):1033. https://doi.org/10.3390/biomedicines9081033
Chicago/Turabian StyleSchoch, Sarah, Vasily Sen, Walburgis Brenner, Andrea Hartwig, and Beate Köberle. 2021. "In Vitro Nephrotoxicity Studies of Established and Experimental Platinum-Based Compounds" Biomedicines 9, no. 8: 1033. https://doi.org/10.3390/biomedicines9081033