
Emerging Role of C5 Complement Pathway in Peripheral Neuropathies: Current Treatments and Future Perspectives

 ,
,  ,
,  ,
,  , ,
, ,
Abstract
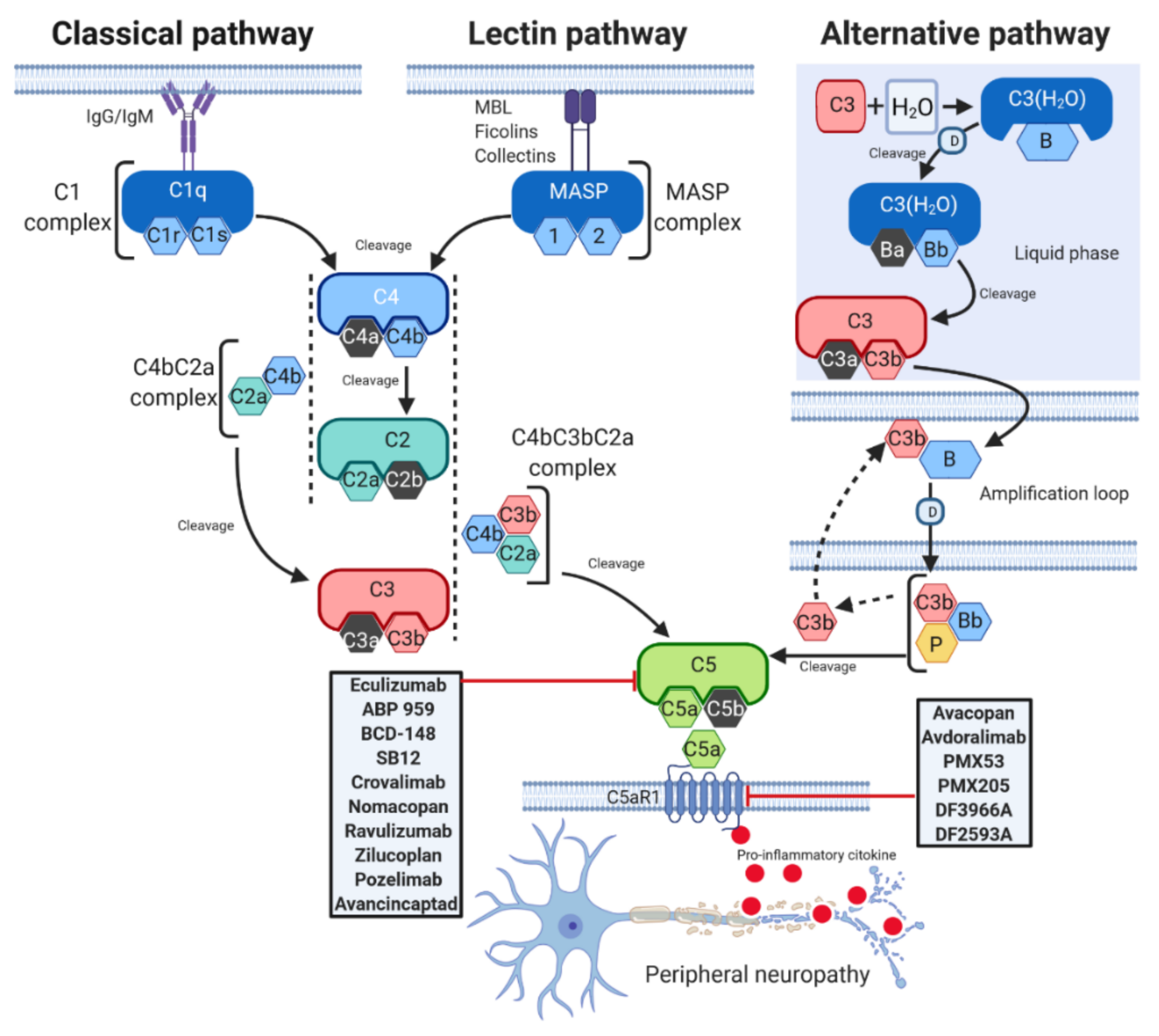
:1. Introduction
2. Complement Pathways
3. Peripheral Neuropathies and C5a/C5aR1 Axis
4. Disease of the Peripheral Nervous System (PNS)
4.1. Guillain-Barré Syndrome
4.2. Chronic Inflammatory Demyelinating Polyradiculoneuropathy
4.3. Familial Amyloid Polyneuropathy
4.4. Chemotherapy-Induced Peripheral Neuropathy
5. C5 Component Targeted Therapies
6. Targeting C5
7. Targeting C5aR
8. Allosteric C5a Receptor Inhibitors
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Nesargikar, P.; Spiller, B.; Chavez, R. The complement system: History, pathways, cascade and inhibitors. Eur. J. Microbiol. Immunol. 2012, 2, 103–111. [Google Scholar] [CrossRef]
- Merle, N.S.; Church, S.E.; Fremeaux-Bacchi, V.; Roumenina, L.T. Complement System Part I—Molecular Mechanisms of Activation and Regulation. Front. Immunol. 2015, 6, 262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ricklin, D.; Lambris, J.D. Complement in immune and inflammatory disorders: Pathophysiological mechanisms. J. Immunol. 2013, 190, 3831–3838. [Google Scholar] [CrossRef] [Green Version]
- Manthey, H.D.; Woodruff, T.M.; Taylor, S.M.; Monk, P.N. Complement component 5a (C5a). Int. J. Biochem. Cell Biol. 2009, 41, 2114–2117. [Google Scholar] [CrossRef]
- Guo, R.F.; Ward, P.A. Role of C5a in inflammatory responses. Annu. Rev. Immunol. 2005, 23, 821–852. [Google Scholar] [CrossRef]
- Heesterbeek, D.A.; Bardoel, B.W.; Parsons, E.S.; Bennett, I.; Ruyken, M.; Doorduijn, D.J.; Gorham, R.D., Jr.; Berends, E.T.; Pyne, A.L.; Hoogenboom, B.W.; et al. Bacterial killing by complement requires membrane attack complex formation via surface-bound C5 convertases. EMBO J. 2019, 38, e99852. [Google Scholar] [CrossRef] [PubMed]
- Fritzinger, D.C.; Benjamin, D.E. The Complement System in Neuropathic and Postoperative Pain. Open Pain J. 2016, 9, 26–37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quadros, A.U.; Cunha, T.M. C5a and pain development: An old molecule, a new target. Pharmacol. Res. 2016, 112, 58–67. [Google Scholar] [CrossRef] [PubMed]
- Merle, N.S.; Noe, R.; Halbwachs-Mecarelli, L.; Fremeaux-Bacchi, V.; Roumenina, L.T. Complement System Part II: Role in Immunity. Front. Immunol. 2015, 6, 257. [Google Scholar] [CrossRef] [Green Version]
- Mamidi, S.; Hone, S.; Kirschfink, M. The complement system in cancer: Ambivalence between tumour destruction and promotion. Immunobiology 2017, 222, 45–54. [Google Scholar] [CrossRef]
- Cedzynski, M.; Swierzko, A.S. Components of the Lectin Pathway of Complement in Haematologic Malignancies. Cancers 2020, 12, 1792. [Google Scholar] [CrossRef] [PubMed]
- Mortensen, S.A.; Sander, B.; Jensen, R.K.; Pedersen, J.S.; Golas, M.M.; Thiel, S.; Andersen, G.R. Models of the complement C1 complex. Proc. Natl. Acad. Sci. USA 2018, 115, E3866. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaboriaud, C.; Thielens, N.M.; Gregory, L.A.; Rossi, V.; Fontecilla-Camps, J.C.; Arlaud, G.J. Structure and activation of the C1 complex of complement: Unraveling the puzzle. Trends Immunol. 2004, 25, 368–373. [Google Scholar] [CrossRef]
- Zwarthoff, S.A.; Berends, E.T.M.; Mol, S.; Ruyken, M.; Aerts, P.C.; Jozsi, M.; de Haas, C.J.C.; Rooijakkers, S.H.M.; Gorham, R.D., Jr. Functional Characterization of Alternative and Classical Pathway C3/C5 Convertase Activity and Inhibition Using Purified Models. Front. Immunol. 2018, 9, 1691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kjaer, T.R.; Jensen, L.; Hansen, A.; Dani, R.; Jensenius, J.C.; Dobo, J.; Gal, P.; Thiel, S. Oligomerization of Mannan-binding Lectin Dictates Binding Properties and Complement Activation. Scand. J. Immunol. 2016, 84, 12–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, M.; Iwaki, D.; Kanno, K.; Ishida, Y.; Xiong, J.; Matsushita, M.; Endo, Y.; Miura, S.; Ishii, N.; Sugamura, K.; et al. Mannose-binding lectin (MBL)-associated serine protease (MASP)-1 contributes to activation of the lectin complement pathway. J. Immunol. 2008, 180, 6132–6138. [Google Scholar] [CrossRef]
- Mortensen, S.; Kidmose, R.T.; Petersen, S.V.; Szilagyi, A.; Prohaszka, Z.; Andersen, G.R. Structural Basis for the Function of Complement Component C4 within the Classical and Lectin Pathways of Complement. J. Immunol. 2015, 194, 5488–5496. [Google Scholar] [CrossRef] [Green Version]
- Thurman, J.M.; Holers, V.M. The central role of the alternative complement pathway in human disease. J. Immunol. 2006, 176, 1305–1310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Z.A.; Pellarin, R.; Fischer, L.; Sali, A.; Nilges, M.; Barlow, P.N.; Rappsilber, J. Structure of Complement C3(H2O) Revealed By Quantitative Cross-Linking/Mass Spectrometry And Modeling. Mol. Cell Proteom. 2016, 15, 2730–2743. [Google Scholar] [CrossRef] [Green Version]
- Hernandez, M.X.; Namiranian, P.; Nguyen, E.; Fonseca, M.I.; Tenner, A.J. C5a Increases the Injury to Primary Neurons Elicited by Fibrillar Amyloid Beta. ASN Neuro 2017, 9, 1759091416687871. [Google Scholar] [CrossRef]
- Ramaglia, V.; Daha, M.R.; Baas, F. The complement system in the peripheral nerve: Friend or foe? Mol. Immunol. 2008, 45, 3865–3877. [Google Scholar] [CrossRef] [PubMed]
- Dalakas, M.C.; Alexopoulos, H.; Spaeth, P.J. Complement in neurological disorders and emerging complement-targeted therapeutics. Nat. Rev. Neurol. 2020, 16, 601–617. [Google Scholar] [CrossRef] [PubMed]
- Hughes, R.A. Peripheral neuropathy. BMJ 2002, 324, 466–469. [Google Scholar] [CrossRef]
- Costigan, M.; Scholz, J.; Woolf, C.J. Neuropathic pain: A maladaptive response of the nervous system to damage. Annu. Rev. Neurosci. 2009, 32, 1–32. [Google Scholar] [CrossRef] [Green Version]
- Reichling, D.B.; Levine, J.D. Pain and death: Neurodegenerative disease mechanisms in the nociceptor. Ann. Neurol. 2011, 69, 13–21. [Google Scholar] [CrossRef] [PubMed]
- Jin, H.W.; Flatters, S.J.; Xiao, W.H.; Mulhern, H.L.; Bennett, G.J. Prevention of paclitaxel-evoked painful peripheral neuropathy by acetyl-L-carnitine: Effects on axonal mitochondria, sensory nerve fiber terminal arbors, and cutaneous Langerhans cells. Exp. Neurol. 2008, 210, 229–237. [Google Scholar] [CrossRef] [Green Version]
- Head, K.A. Peripheral neuropathy: Pathogenic mechanisms and alternative therapies. Altern. Med. Rev. 2006, 11, 294–329. [Google Scholar] [PubMed]
- Hanewinckel, R.; Ikram, M.A.; Van Doorn, P.A. Peripheral neuropathies. Handb. Clin. Neurol. 2016, 138, 263–282. [Google Scholar] [CrossRef]
- Colloca, L.; Ludman, T.; Bouhassira, D.; Baron, R.; Dickenson, A.H.; Yarnitsky, D.; Freeman, R.; Truini, A.; Attal, N.; Finnerup, N.B.; et al. Neuropathic pain. Nat. Rev. Dis. Primers 2017, 3, 170002. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Girach, A.; Julian, T.H.; Varrassi, G.; Paladini, A.; Vadalouka, A.; Zis, P. Quality of Life in Painful Peripheral Neuropathies: A Systematic Review. Pain Res. Manag. 2019, 2019, 2091960. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liampas, A.; Rekatsina, M.; Vadalouca, A.; Paladini, A.; Varrassi, G.; Zis, P. Non-Pharmacological Management of Painful Peripheral Neuropathies: A Systematic Review. Adv. Ther. 2020, 37, 4096–4106. [Google Scholar] [CrossRef]
- Liampas, A.; Rekatsina, M.; Vadalouca, A.; Paladini, A.; Varrassi, G.; Zis, P. Pharmacological Management of Painful Peripheral Neuropathies: A Systematic Review. Pain Ther. 2020, 1–14. [Google Scholar] [CrossRef]
- Nabizadeh, J.A.; Manthey, H.D.; Panagides, N.; Steyn, F.J.; Lee, J.D.; Li, X.X.; Akhir, F.N.M.; Chen, W.; Boyle, G.M.; Taylor, S.M.; et al. C5a receptors C5aR1 and C5aR2 mediate opposing pathologies in a mouse model of melanoma. FASEB J. 2019, 33, 11060–11071. [Google Scholar] [CrossRef] [Green Version]
- Allegretti, M.; Moriconi, A.; Beccari, A.R.; Di Bitondo, R.; Bizzarri, C.; Bertini, R.; Colotta, F. Targeting C5a: Recent advances in drug discovery. Curr. Med. Chem. 2005, 12, 217–236. [Google Scholar] [CrossRef] [PubMed]
- Li, X.X.; Lee, J.D.; Kemper, C.; Woodruff, T.M. The Complement Receptor C5aR2: A Powerful Modulator of Innate and Adaptive Immunity. J. Immunol. 2019, 202, 3339–3348. [Google Scholar] [CrossRef]
- Peng, Q.; Li, K.; Sacks, S.H.; Zhou, W. The role of anaphylatoxins C3a and C5a in regulating innate and adaptive immune responses. Inflamm. Allergy Drug Targets 2009, 8, 236–246. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Garstka, M.A.; Li, K. The Controversial C5a Receptor C5aR2: Its Role in Health and Disease. J. Immunol. Res. 2017, 2017, 8193932. [Google Scholar] [CrossRef] [Green Version]
- Croker, D.E.; Monk, P.N.; Halai, R.; Kaeslin, G.; Schofield, Z.; Wu, M.C.; Clark, R.J.; Blaskovich, M.A.; Morikis, D.; Floudas, C.A.; et al. Discovery of functionally selective C5aR2 ligands: Novel modulators of C5a signalling. Immunol. Cell Biol. 2016, 94, 787–795. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiese, A.V.; Ender, F.; Quell, K.M.; Antoniou, K.; Vollbrandt, T.; Konig, P.; Kohl, J.; Laumonnier, Y. The C5a/C5aR1 axis controls the development of experimental allergic asthma independent of LysM-expressing pulmonary immune cells. PLoS ONE 2017, 12, e0184956. [Google Scholar] [CrossRef] [Green Version]
- Moriconi, A.; Cunha, T.M.; Souza, G.R.; Lopes, A.H.; Cunha, F.Q.; Carneiro, V.L.; Pinto, L.G.; Brandolini, L.; Aramini, A.; Bizzarri, C.; et al. Targeting the minor pocket of C5aR for the rational design of an oral allosteric inhibitor for inflammatory and neuropathic pain relief. Proc. Natl. Acad. Sci. USA 2014, 111, 16937–16942. [Google Scholar] [CrossRef] [Green Version]
- Shutov, L.P.; Warwick, C.A.; Shi, X.; Gnanasekaran, A.; Shepherd, A.J.; Mohapatra, D.P.; Woodruff, T.M.; Clark, J.D.; Usachev, Y.M. The Complement System Component C5a Produces Thermal Hyperalgesia via Macrophage-to-Nociceptor Signaling That Requires NGF and TRPV1. J. Neurosci. 2016, 36, 5055–5070. [Google Scholar] [CrossRef]
- Griffin, R.S.; Costigan, M.; Brenner, G.J.; Ma, C.H.; Scholz, J.; Moss, A.; Allchorne, A.J.; Stahl, G.L.; Woolf, C.J. Complement induction in spinal cord microglia results in anaphylatoxin C5a-mediated pain hypersensitivity. J. Neurosci. 2007, 27, 8699–8708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, D.Y.; Li, X.; Shi, X.; Sun, Y.; Sahbaie, P.; Li, W.W.; Clark, J.D. The complement component C5a receptor mediates pain and inflammation in a postsurgical pain model. Pain 2012, 153, 366–372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clark, J.D.; Qiao, Y.; Li, X.; Shi, X.; Angst, M.S.; Yeomans, D.C. Blockade of the complement C5a receptor reduces incisional allodynia, edema, and cytokine expression. Anesthesiology 2006, 104, 1274–1282. [Google Scholar] [CrossRef]
- Ting, E.; Guerrero, A.T.; Cunha, T.M.; Verri, W.A., Jr.; Taylor, S.M.; Woodruff, T.M.; Cunha, F.Q.; Ferreira, S.H. Role of complement C5a in mechanical inflammatory hypernociception: Potential use of C5a receptor antagonists to control inflammatory pain. Br. J. Pharmacol. 2008, 153, 1043–1053. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hafer-Macko, C.; Hsieh, S.T.; Li, C.Y.; Ho, T.W.; Sheikh, K.; Cornblath, D.R.; McKhann, G.M.; Asbury, A.K.; Griffin, J.W. Acute motor axonal neuropathy: An antibody-mediated attack on axolemma. Ann. Neurol. 1996, 40, 635–644. [Google Scholar] [CrossRef]
- Kuwabara, S.; Yuki, N. Axonal Guillain-Barre syndrome: Concepts and controversies. Lancet Neurol. 2013, 12, 1180–1188. [Google Scholar] [CrossRef]
- Jasti, A.K.; Selmi, C.; Sarmiento-Monroy, J.C.; Vega, D.A.; Anaya, J.M.; Gershwin, M.E. Guillain-Barre syndrome: Causes, immunopathogenic mechanisms and treatment. Expert Rev. Clin. Immunol. 2016, 12, 1175–1189. [Google Scholar] [CrossRef]
- McGrogan, A.; Madle, G.C.; Seaman, H.E.; de Vries, C.S. The epidemiology of Guillain-Barre syndrome worldwide. A systematic literature review. Neuroepidemiology 2009, 32, 150–163. [Google Scholar] [CrossRef]
- Jacobs, B.C.; O’Hanlon, G.M.; Bullens, R.W.; Veitch, J.; Plomp, J.J.; Willison, H.J. Immunoglobulins inhibit pathophysiological effects of anti-GQ1b-positive sera at motor nerve terminals through inhibition of antibody binding. Brain 2003, 126, 2220–2234. [Google Scholar] [CrossRef]
- Fokke, C.; van den Berg, B.; Drenthen, J.; Walgaard, C.; van Doorn, P.A.; Jacobs, B.C. Diagnosis of Guillain-Barre syndrome and validation of Brighton criteria. Brain 2014, 137, 33–43. [Google Scholar] [CrossRef] [Green Version]
- Schaller, B.; Radziwill, A.J.; Steck, A.J. Successful treatment of Guillain-Barre syndrome with combined administration of interferon-beta-1a and intravenous immunoglobulin. Eur. Neurol. 2001, 46, 167–168. [Google Scholar] [CrossRef]
- Raphael, J.C.; Chevret, S.; Hughes, R.A.; Annane, D. Plasma exchange for Guillain-Barre syndrome. Cochrane Database Syst. Rev. 2002, 7, CD001798. [Google Scholar] [CrossRef]
- Meyer zu Horste, G.; Hartung, H.P.; Kieseier, B.C. From bench to bedside--experimental rationale for immune-specific therapies in the inflamed peripheral nerve. Nat. Clin. Pract. Neurol. 2007, 3, 198–211. [Google Scholar] [CrossRef] [PubMed]
- O’Hanlon, G.M.; Plomp, J.J.; Chakrabarti, M.; Morrison, I.; Wagner, E.R.; Goodyear, C.S.; Yin, X.; Trapp, B.D.; Conner, J.; Molenaar, P.C.; et al. Anti-GQ1b ganglioside antibodies mediate complement-dependent destruction of the motor nerve terminal. Brain 2001, 124, 893–906. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Halstead, S.K.; O’Hanlon, G.M.; Humphreys, P.D.; Morrison, D.B.; Morgan, B.P.; Todd, A.J.; Plomp, J.J.; Willison, H.J. Anti-disialoside antibodies kill perisynaptic Schwann cells and damage motor nerve terminals via membrane attack complex in a murine model of neuropathy. Brain 2004, 127, 2109–2123. [Google Scholar] [CrossRef]
- Halstead, S.K.; Humphreys, P.D.; Goodfellow, J.A.; Wagner, E.R.; Smith, R.A.; Willison, H.J. Complement inhibition abrogates nerve terminal injury in Miller Fisher syndrome. Ann. Neurol. 2005, 58, 203–210. [Google Scholar] [CrossRef]
- Halstead, S.K.; Zitman, F.M.; Humphreys, P.D.; Greenshields, K.; Verschuuren, J.J.; Jacobs, B.C.; Rother, R.P.; Plomp, J.J.; Willison, H.J. Eculizumab prevents anti-ganglioside antibody-mediated neuropathy in a murine model. Brain 2008, 131, 1197–1208. [Google Scholar] [CrossRef] [Green Version]
- McGonigal, R.; Cunningham, M.E.; Yao, D.; Barrie, J.A.; Sankaranarayanan, S.; Fewou, S.N.; Furukawa, K.; Yednock, T.A.; Willison, H.J. C1q-targeted inhibition of the classical complement pathway prevents injury in a novel mouse model of acute motor axonal neuropathy. Acta Neuropathol. Commun. 2016, 4, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Dalakas, M.C. Medscape. Advances in the diagnosis, pathogenesis and treatment of CIDP. Nat. Rev. Neurol. 2011, 7, 507–517. [Google Scholar] [CrossRef]
- Broers, M.C.; Bunschoten, C.; Nieboer, D.; Lingsma, H.F.; Jacobs, B.C. Incidence and Prevalence of Chronic Inflammatory Demyelinating Polyradiculoneuropathy: A Systematic Review and Meta-Analysis. Neuroepidemiology 2019, 52, 161–172. [Google Scholar] [CrossRef] [PubMed]
- Koller, H.; Kieseier, B.C.; Jander, S.; Hartung, H.P. Chronic inflammatory demyelinating polyneuropathy. N. Engl. J. Med. 2005, 352, 1343–1356. [Google Scholar] [CrossRef] [Green Version]
- Mathey, E.K.; Park, S.B.; Hughes, R.A.; Pollard, J.D.; Armati, P.J.; Barnett, M.H.; Taylor, B.V.; Dyck, P.J.; Kiernan, M.C.; Lin, C.S. Chronic inflammatory demyelinating polyradiculoneuropathy: From pathology to phenotype. J. Neurol. Neurosurg. Psychiatry 2015, 86, 973–985. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dalakas, M.C.; Engel, W.K. Immunoglobulin and complement deposits in nerves of patients with chronic relapsing polyneuropathy. Arch. Neurol. 1980, 37, 637–640. [Google Scholar] [CrossRef] [PubMed]
- Hays, A.P.; Lee, S.S.; Latov, N. Immune reactive C3d on the surface of myelin sheaths in neuropathy. J. Neuroimmunol. 1988, 18, 231–244. [Google Scholar] [CrossRef]
- Quast, I.; Keller, C.W.; Hiepe, F.; Tackenberg, B.; Lunemann, J.D. Terminal complement activation is increased and associated with disease severity in CIDP. Ann. Clin. Transl. Neurol. 2016, 3, 730–735. [Google Scholar] [CrossRef]
- Cakar, A.; Durmus-Tekce, H.; Parman, Y. Familial Amyloid Polyneuropathy. Arch. Neurol. 2019, 56, 150. [Google Scholar] [CrossRef]
- Kato-Motozaki, Y.; Ono, K.; Shima, K.; Morinaga, A.; Machiya, T.; Nozaki, I.; Shibata-Hamaguchi, A.; Furukawa, Y.; Yanase, D.; Ishida, C.; et al. Epidemiology of familial amyloid polyneuropathy in Japan: Identification of a novel endemic focus. J. Neurol. Sci. 2008, 270, 133–140. [Google Scholar] [CrossRef]
- Ando, Y.; Nakamura, M.; Araki, S. Transthyretin-related familial amyloidotic polyneuropathy. Arch. Neurol. 2005, 62, 1057–1062. [Google Scholar] [CrossRef] [Green Version]
- Plante-Bordeneuve, V.; Said, G. Familial amyloid polyneuropathy. Lancet Neurol. 2011, 10, 1086–1097. [Google Scholar] [CrossRef]
- Rowczenio, D.; Quarta, C.C.; Fontana, M.; Whelan, C.J.; Martinez-Naharro, A.; Trojer, H.; Baginska, A.; Ferguson, S.M.; Gilbertson, J.; Rezk, T.; et al. Analysis of the TTR gene in the investigation of amyloidosis: A 25-year single UK center experience. Hum. Mutat. 2019, 40, 90–96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manganelli, F.; Fabrizi, G.M.; Luigetti, M.; Mandich, P.; Mazzeo, A.; Pareyson, D. Hereditary transthyretin amyloidosis overview. Neurol. Sci. 2020. [Google Scholar] [CrossRef] [PubMed]
- Ohmori, H.; Ando, Y.; Makita, Y.; Onouchi, Y.; Nakajima, T.; Saraiva, M.J.; Terazaki, H.; Suhr, O.; Sobue, G.; Nakamura, M.; et al. Common origin of the Val30Met mutation responsible for the amyloidogenic transthyretin type of familial amyloidotic polyneuropathy. J. Med. Genet. 2004, 41, e51. [Google Scholar] [CrossRef] [Green Version]
- Ando, Y.; Coelho, T.; Berk, J.L.; Cruz, M.W.; Ericzon, B.G.; Ikeda, S.; Lewis, W.D.; Obici, L.; Plante-Bordeneuve, V.; Rapezzi, C.; et al. Guideline of transthyretin-related hereditary amyloidosis for clinicians. Orphanet J. Rare Dis. 2013, 8, 31. [Google Scholar] [CrossRef] [Green Version]
- Benson, M.D. Liver transplantation and transthyretin amyloidosis. Muscle Nerve 2013, 47, 157–162. [Google Scholar] [CrossRef]
- Hafer-Macko, C.E.; Dyck, P.J.; Koski, C.L. Complement activation in acquired and hereditary amyloid neuropathy. J. Peripher. Nerv. Syst. 2000, 5, 131–139. [Google Scholar] [CrossRef]
- Fonseca, M.I.; Zhou, J.; Botto, M.; Tenner, A.J. Absence of C1q leads to less neuropathology in transgenic mouse models of Alzheimer’s disease. J. Neurosci. 2004, 24, 6457–6465. [Google Scholar] [CrossRef] [Green Version]
- Pisalyaput, K.; Tenner, A.J. Complement component C1q inhibits beta-amyloid- and serum amyloid P-induced neurotoxicity via caspase- and calpain-independent mechanisms. J. Neurochem. 2008, 104, 696–707. [Google Scholar] [CrossRef] [PubMed]
- Galvan, M.D.; Foreman, D.B.; Zeng, E.; Tan, J.C.; Bohlson, S.S. Complement component C1q regulates macrophage expression of Mer tyrosine kinase to promote clearance of apoptotic cells. J. Immunol. 2012, 188, 3716–3723. [Google Scholar] [CrossRef] [Green Version]
- Fonseca, M.I.; Chu, S.H.; Berci, A.M.; Benoit, M.E.; Peters, D.G.; Kimura, Y.; Tenner, A.J. Contribution of complement activation pathways to neuropathology differs among mouse models of Alzheimer’s disease. J. Neuroinflamm. 2011, 8, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Quasthoff, S.; Hartung, H.P. Chemotherapy-induced peripheral neuropathy. J. Neurol. 2002, 249, 9–17. [Google Scholar] [CrossRef]
- Pike, C.T.; Birnbaum, H.G.; Muehlenbein, C.E.; Pohl, G.M.; Natale, R.B. Healthcare costs and workloss burden of patients with chemotherapy-associated peripheral neuropathy in breast, ovarian, head and neck, and nonsmall cell lung cancer. Chemother. Res. Pract. 2012, 2012, 913848. [Google Scholar] [CrossRef]
- Balayssac, D.; Ferrier, J.; Descoeur, J.; Ling, B.; Pezet, D.; Eschalier, A.; Authier, N. Chemotherapy-induced peripheral neuropathies: From clinical relevance to preclinical evidence. Expert Opin. Drug Saf. 2011, 10, 407–417. [Google Scholar] [CrossRef]
- Kolb, N.A.; Smith, A.G.; Singleton, J.R.; Beck, S.L.; Stoddard, G.J.; Brown, S.; Mooney, K. The Association of Chemotherapy-Induced Peripheral Neuropathy Symptoms and the Risk of Falling. JAMA Neurol. 2016, 73, 860–866. [Google Scholar] [CrossRef] [Green Version]
- Salat, K. Chemotherapy-induced peripheral neuropathy: Part 1-current state of knowledge and perspectives for pharmacotherapy. Pharmacol. Rep. 2020, 72, 486–507. [Google Scholar] [CrossRef] [PubMed]
- Salat, K. Chemotherapy-induced peripheral neuropathy-part 2: Focus on the prevention of oxaliplatin-induced neurotoxicity. Pharmacol. Rep. 2020, 72, 508–527. [Google Scholar] [CrossRef] [PubMed]
- Lees, J.G.; Makker, P.G.; Tonkin, R.S.; Abdulla, M.; Park, S.B.; Goldstein, D.; Moalem-Taylor, G. Immune-mediated processes implicated in chemotherapy-induced peripheral neuropathy. Eur. J. Cancer 2017, 73, 22–29. [Google Scholar] [CrossRef] [PubMed]
- Brandolini, L.; d’Angelo, M.; Antonosante, A.; Allegretti, M.; Cimini, A. Chemokine Signaling in Chemotherapy-Induced Neuropathic Pain. Int. J. Mol. Sci. 2019, 20, 2904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, J.; Zhang, L.; Xie, M.; Li, Y.; Huang, P.; Saunders, T.L.; Fox, D.A.; Rosenquist, R.; Lin, F. Role of Complement in a Rat Model of Paclitaxel-Induced Peripheral Neuropathy. J. Immunol. 2018, 200, 4094–4101. [Google Scholar] [CrossRef] [PubMed]
- Jang, J.H.; Clark, D.J.; Li, X.; Yorek, M.S.; Usachev, Y.M.; Brennan, T.J. Nociceptive sensitization by complement C5a and C3a in mouse. Pain 2010, 148, 343–352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jang, J.H.; Liang, D.; Kido, K.; Sun, Y.; Clark, D.J.; Brennan, T.J. Increased local concentration of complement C5a contributes to incisional pain in mice. J. Neuroinflamm. 2011, 8, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Ricklin, D.; Lambris, J.D. New milestones ahead in complement-targeted therapy. Semin. Immunol. 2016, 28, 208–222. [Google Scholar] [CrossRef] [Green Version]
- Matis, L.A.; Rollins, S.A. Complement-specific antibodies: Designing novel anti-inflammatories. Nat. Med. 1995, 1, 839–842. [Google Scholar] [CrossRef]
- Schatz-Jakobsen, J.A.; Zhang, Y.; Johnson, K.; Neill, A.; Sheridan, D.; Andersen, G.R. Structural Basis for Eculizumab-Mediated Inhibition of the Complement Terminal Pathway. J. Immunol. 2016, 197, 337–344. [Google Scholar] [CrossRef] [Green Version]
- Horiuchi, T.; Tsukamoto, H. Complement-targeted therapy: Development of C5- and C5a-targeted inhibition. Inflamm. Regen. 2016, 36, 1–5. [Google Scholar] [CrossRef] [Green Version]
- US Food and Drug Administration. Soliris® (Eculizumab) [Prescribing Information]; Alexion Pharmaceuticals, Inc.: Cheshire, CT, USA, 2014. [Google Scholar]
- Dmytrijuk, A.; Robie-Suh, K.; Cohen, M.H.; Rieves, D.; Weiss, K.; Pazdur, R. FDA report: Eculizumab (Soliris) for the treatment of patients with paroxysmal nocturnal hemoglobinuria. Oncologist 2008, 13, 993–1000. [Google Scholar] [CrossRef] [Green Version]
- Palma, L.M.; Langman, C.B. Critical appraisal of eculizumab for atypical hemolytic uremic syndrome. J. Blood Med. 2016, 7, 39–72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Misawa, S.; Kuwabara, S.; Sato, Y.; Yamaguchi, N.; Nagashima, K.; Katayama, K.; Sekiguchi, Y.; Iwai, Y.; Amino, H.; Suichi, T.; et al. Safety and efficacy of eculizumab in Guillain-Barre syndrome: A multicentre, double-blind, randomised phase 2 trial. Lancet Neurol. 2018, 17, 519–529. [Google Scholar] [CrossRef]
- Pittock, S.J.; Berthele, A.; Fujihara, K.; Kim, H.J.; Levy, M.; Palace, J.; Nakashima, I.; Terzi, M.; Totolyan, N.; Viswanathan, S.; et al. Eculizumab in Aquaporin-4-Positive Neuromyelitis Optica Spectrum Disorder. N. Engl. J. Med. 2019, 381, 614–625. [Google Scholar] [CrossRef] [PubMed]
- Tan, E.K.; Bentall, A.; Dean, P.G.; Shaheen, M.F.; Stegall, M.D.; Schinstock, C.A. Use of Eculizumab for Active Antibody-mediated Rejection That Occurs Early Post-kidney Transplantation: A Consecutive Series of 15 Cases. Transplantation 2019, 103, 2397–2404. [Google Scholar] [CrossRef]
- De Holanda, M.I.; Porto, L.C.; Wagner, T.; Christiani, L.F.; Palma, L.M.P. Use of eculizumab in a systemic lupus erythemathosus patient presenting thrombotic microangiopathy and heterozygous deletion in CFHR1-CFHR3. A case report and systematic review. Clin. Rheumatol. 2017, 36, 2859–2867. [Google Scholar] [CrossRef] [PubMed]
- Schulte-Kemna, L.; Reister, B.; Bettac, L.; Ludwig, U.; Furst, D.; Mytilineos, J.; Bergmann, C.; van Erp, R.; Schroppel, B. Eculizumab in chemotherapy-induced thrombotic microangiopathy. Clin. Nephrol. Case Stud. 2020, 8, 25–32. [Google Scholar] [CrossRef] [PubMed]
- Dhillon, S. Eculizumab: A Review in Generalized Myasthenia Gravis. Drugs 2018, 78, 367–376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishimura, J.I. Antibody therapy for paroxysmal nocturnal hemoglobinuria. Rinsho Ketsueki 2020, 61, 929–936. [Google Scholar] [CrossRef] [PubMed]
- Roth, A.; Nishimura, J.I.; Nagy, Z.; Gaal-Weisinger, J.; Panse, J.; Yoon, S.S.; Egyed, M.; Ichikawa, S.; Ito, Y.; Kim, J.S.; et al. The complement C5 inhibitor crovalimab in paroxysmal nocturnal hemoglobinuria. Blood 2020, 135, 912–920. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schols, S.; Nunn, M.A.; Mackie, I.; Weston-Davies, W.; Nishimura, J.I.; Kanakura, Y.; Blijlevens, N.; Muus, P.; Langemeijer, S. Successful treatment of a PNH patient non-responsive to eculizumab with the novel complement C5 inhibitor coversin (nomacopan). Br. J. Haematol. 2020, 188, 334–337. [Google Scholar] [CrossRef]
- Rondeau, E.; Scully, M.; Ariceta, G.; Barbour, T.; Cataland, S.; Heyne, N.; Miyakawa, Y.; Ortiz, S.; Swenson, E.; Vallee, M.; et al. The long-acting C5 inhibitor, Ravulizumab, is effective and safe in adult patients with atypical hemolytic uremic syndrome naive to complement inhibitor treatment. Kidney Int. 2020, 97, 1287–1296. [Google Scholar] [CrossRef]
- Howard, J.F., Jr.; Nowak, R.J.; Wolfe, G.I.; Freimer, M.L.; Vu, T.H.; Hinton, J.L.; Benatar, M.; Duda, P.W.; MacDougall, J.E.; Farzaneh-Far, R.; et al. Clinical Effects of the Self-administered Subcutaneous Complement Inhibitor Zilucoplan in Patients With Moderate to Severe Generalized Myasthenia Gravis: Results of a Phase 2 Randomized, Double-Blind, Placebo-Controlled, Multicenter Clinical Trial. JAMA Neurol. 2020, 77, 582–592. [Google Scholar] [CrossRef]
- Latuszek, A.; Liu, Y.; Olsen, O.; Foster, R.; Cao, M.; Lovric, I.; Yuan, M.; Liu, N.; Chen, H.; Zhang, Q.; et al. Inhibition of complement pathway activation with Pozelimab, a fully human antibody to complement component C5. PLoS ONE 2020, 15, e0231892. [Google Scholar] [CrossRef]
- Jaffe, G.J.; Westby, K.; Csaky, K.G.; Mones, J.; Pearlman, J.A.; Patel, S.S.; Joondeph, B.C.; Randolph, J.; Masonson, H.; Rezaei, K.A. C5 Inhibitor Avacincaptad Pegol for Geographic Atrophy Due to Age-Related Macular Degeneration: A Randomized Pivotal Phase 2/3 Trial. Ophthalmology 2020, 128, 576–586. [Google Scholar] [CrossRef]
- Merkel, P.A.; Jayne, D.R.; Wang, C.; Hillson, J.; Bekker, P. Evaluation of the Safety and Efficacy of Avacopan, a C5a Receptor Inhibitor, in Patients With Antineutrophil Cytoplasmic Antibody-Associated Vasculitis Treated Concomitantly With Rituximab or Cyclophosphamide/Azathioprine: Protocol for a Randomized, Double-Blind, Active-Controlled, Phase 3 Trial. JMIR Res. Protoc. 2020, 9, e16664. [Google Scholar] [CrossRef] [Green Version]
- World Health Organization. International Nonproprietary Names for Pharmaceutical Substances (INN): Proposed INN: List 121; No. 2; WHO Drug Inf.: Geneva, Switzerland, 2019; Volume 33. [Google Scholar]
- Llaudo, I.; Fribourg, M.; Medof, M.E.; Conde, P.; Ochando, J.; Heeger, P.S. C5aR1 regulates migration of suppressive myeloid cells required for costimulatory blockade-induced murine allograft survival. Am. J. Transplant. 2019, 19, 633–645. [Google Scholar] [CrossRef]
- Ghouse, S.M.; Vadrevu, S.K.; Manne, S.; Reese, B.; Patel, J.; Patel, B.; Silwal, A.; Lodhi, N.; Paterson, Y.; Srivastava, S.K.; et al. Therapeutic Targeting of Vasculature in the Premetastatic and Metastatic Niches Reduces Lung Metastasis. J. Immunol. 2020, 204, 990–1000. [Google Scholar] [CrossRef]
- Woodruff, T.M.; Nandakumar, K.S.; Tedesco, F. Inhibiting the C5-C5a receptor axis. Mol. Immunol. 2011, 48, 1631–1642. [Google Scholar] [CrossRef] [PubMed]
- Harris, C.L. Expanding horizons in complement drug discovery: Challenges and emerging strategies. Semin. Immunopathol. 2018, 40, 125–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ricklin, D.; Lambris, J.D. Complement-targeted therapeutics. Nat. Biotechnol. 2007, 25, 1265–1275. [Google Scholar] [CrossRef]
- Vignesh, P.; Rawat, A.; Singh, S. An Update on the Use of Immunomodulators in Primary Immunodeficiencies. Clin. Rev. Allergy Immunol. 2017, 52, 287–303. [Google Scholar] [CrossRef] [PubMed]
- McNamara, L.A.; Topaz, N.; Wang, X.; Hariri, S.; Fox, L.; MacNeil, J.R. High Risk for Invasive Meningococcal Disease Among Patients Receiving Eculizumab (Soliris) Despite Receipt of Meningococcal Vaccine. MMWR. Morb. Mortal. Wkly. Rep. 2017, 66, 734–737. [Google Scholar] [CrossRef] [Green Version]
- Langereis, J.D.; van den Broek, B.; Franssen, S.; Joosten, I.; Blijlevens, N.M.A.; de Jonge, M.I.; Langemeijer, S. Eculizumab impairs Neisseria meningitidis serogroup B killing in whole blood despite 4CMenB vaccination of PNH patients. Blood Adv. 2020, 4, 3615–3620. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Kim, H.R.; Deepak, R.; Wang, L.; Chung, K.Y.; Fan, H.; Wei, Z.; Zhang, C. Orthosteric and allosteric action of the C5a receptor antagonists. Nat. Struct. Mol. Biol. 2018, 25, 472–481. [Google Scholar] [CrossRef]
- Kumar, V.; Lee, J.D.; Clark, R.J.; Noakes, P.G.; Taylor, S.M.; Woodruff, T.M. Preclinical Pharmacokinetics of Complement C5a Receptor Antagonists PMX53 and PMX205 in Mice. ACS Omega 2020, 5, 2345–2354. [Google Scholar] [CrossRef] [Green Version]
- Kohl, J. Drug evaluation: The C5a receptor antagonist PMX-53. Curr. Opin. Mol. Ther. 2006, 8, 529–538. [Google Scholar] [PubMed]
- Dumitru, A.C.; Deepak, R.; Liu, H.; Koehler, M.; Zhang, C.; Fan, H.; Alsteens, D. Submolecular probing of the complement C5a receptor-ligand binding reveals a cooperative two-site binding mechanism. Commun. Biol. 2020, 3, 1–13. [Google Scholar] [CrossRef]
- Seow, V.; Lim, J.; Cotterell, A.J.; Yau, M.K.; Xu, W.; Lohman, R.J.; Kok, W.M.; Stoermer, M.J.; Sweet, M.J.; Reid, R.C.; et al. Receptor residence time trumps drug-likeness and oral bioavailability in determining efficacy of complement C5a antagonists. Sci. Rep. 2016, 6, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fredslund, F.; Laursen, N.S.; Roversi, P.; Jenner, L.; Oliveira, C.L.; Pedersen, J.S.; Nunn, M.A.; Lea, S.M.; Discipio, R.; Sottrup-Jensen, L.; et al. Structure of and influence of a tick complement inhibitor on human complement component 5. Nat. Immunol. 2008, 9, 753–760. [Google Scholar] [CrossRef] [PubMed]
- Brandolini, L.; Grannonico, M.; Bianchini, G.; Colanardi, A.; Sebastiani, P.; Paladini, A.; Piroli, A.; Allegretti, M.; Varrassi, G.; Di Loreto, S. The Novel C5aR Antagonist DF3016A Protects Neurons Against Ischemic Neuroinflammatory Injury. Neurotox. Res. 2019, 36, 163–174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harris, C.L.; Pouw, R.B.; Kavanagh, D.; Sun, R.; Ricklin, D. Developments in anti-complement therapy; from disease to clinical trial. Mol. Immunol. 2018, 102, 89–119. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Drug | Indication | Trial Phase/Study Type | Recruitment Status | NCT Number |
|---|---|---|---|---|
| Eculizumab, C5-targeting antibody | PNH | Phase 2/3 | Only recruiting Trials are listed | NCT04079257 |
| aHUS-associated multiple organ dysfunction syndrome in hematopoietic stem cell transplant recipients | Phase 2 | NCT03518203 | ||
| Refractory GMG | Phase 3 | NCT03759366 | ||
| COVID-19 | Phase 2 | NCT04346797 | ||
| CHAPLE disease | Prospective Cohort | NCT03950804 | ||
| PNH | Phase 3 | NCT04432584 | ||
| PNH | Phase 3 | NCT03818607 | ||
| PNH | Phase 3 | NCT04434092 | ||
| End Stage Liver Disease | Phase 1 | NCT03468140 | ||
| PNH | Observational Study | NCT01374360 | ||
| aHUS | Observational Study | NCT01522183 | ||
| Neuromyelitis optica spectrum disorder | Phase 2/3 | NCT04155424 | ||
| ABP959, C5-targeting antibody (eculizumab biosimilar) | PNH | Phase 3 | Recruiting | NCT03818607 |
| SB12 C5-targeting antibody (eculizumab biosimilar) | PNH | Phase 3 | Active, not recruiting | NCT04058158 |
| BCD-148, C5-targeting antibody (eculizumab biosimilar) | PNH | Phase 3 | Active, not recruiting | NCT04060264 |
| Healthy subjects | Phase 1 | Completed | NCT04027803 | |
| Nomacopan, C5-targeting protein | PNH, aHUS | Phase 3 | Recruiting | NCT03829449 |
| BP | Phase 2 | Completed | NCT04035733 | |
| PNH | Phase 3 | Completed | NCT03588026 | |
| PNH | Phase 2 | Enrolling by invitation | NCT03427060 | |
| PNH | Phase 2 | Completed | NCT02591862 | |
| AKC | Phase 1/2 | Active, not recruiting | NCT04037891 | |
| Ravulizumab, C5-targeting antibody | COVID-19, thrombotic microangiopathies acute kidney injury | Phase 3 | Only recruiting Trials are listed | NCT04570397 |
| Neuromyelitis optica Neuromyelitis optica spectrum disorder | Phase 3 | NCT04201262 | ||
| TMA | Phase 3 | NCT04557735 | ||
| ALS | Phase 3 | NCT04248465 | ||
| COVID-19 severe pneumonia, acute lung injury, acute respiratory distress syndrome pneumonia, Viral | Phase 3 | NCT04369469 | ||
| TMA | Phase 3 | NCT04543591 | ||
| PNH | Phase 3 | NCT03406507 | ||
| COVID-19 | Phase 4 | NCT04390464 | ||
| Atypical hemolytic-uremic syndrome | Observational Study | NCT01522183 | ||
| PNH | Observational Study | NCT01374360 | ||
| PNH | Phase 3 | NCT04432584 | ||
| Pozelimab, C5-targeting antibody | Healthy volunteer | Phase 1 | Active, not recruiting | NCT04491838 |
| Healthy subjects (in combination with cemdisiran) | Phase 1 | Recruiting | NCT04601844 | |
| CHAPLE | Phase 2/3 | Recruiting | NCT04209634 | |
| Crovalimab, C5-targeting antibody | PNH | Phase 3 | Not yet recruiting | NCT04654468 |
| PNH | Phase 3 | Recruiting | NCT04432584 | |
| PNH | Phase 3 | Recruiting | NCT04434092 | |
| PNH | Phase 1/2 | Active, not recruiting | NCT03157635 | |
| Cemdisiran, C5 targeting RNAi therapeutic | IgAN, Berger disease, glomerulonephritis | Phase 2 | Recruiting | NCT03841448 |
| Healthy | Phase 1 | Recruiting | NCT04601844 | |
| aHUS | Phase 2 | Withdrawn | NCT03303313 | |
| PNH | Phase 1 | Completed | NCT02352493 | |
| TMA | Phase2 | Not yet recruiting | NCT03999840 | |
| IFX-1, C5a targeting antibody | Pyoderma gangrenosum | Phase 2 | Recruiting | NCT03971643 |
| Severe COVID-19 pneumonia | Phase 2/3 | Recruiting | NCT04333420 | |
| GPA, MPA | Phase 2 | Active, not recruiting | NCT03712345 | |
| GPA, MPA | Phase 2 | Active, not recruiting | NCT03895801 | |
| HS | Phase 2 | Completed | NCT03487276 | |
| HS | Phase 2 | Completed | NCT03001622 | |
| Systemic inflammatory response syndrome, C.surgical procedure, cardiac | Phase 2 | Completed | NCT02866825 | |
| Severe sepsis, septic shock | Phase 2 | Completed | NCT02246595 | |
| Drug Safety | Phase 1 | Completed | NCT01319903 | |
| BDB-001, C5a targeting antibody | COVID-19 pneumonia | Phase 2/3 | Recruiting | NCT04449588 |
| Solid tumor | Phase 1 | Recruiting | NCT04196530 | |
| Solid tumor | Phase 1 | Recruiting | NCT03486301 | |
| Solid tumor, pancreatic cancer, virus associated tumors, non-small cell lung cancer, melanoma, bladdder cancer, triple negative breast cancer | Phase 2 | Not yet recruiting | NCT03915678 | |
| ALXN1007, C5a targeting antibody | Antiphospholipid (aPL)-positive | Phase 2 | Terminated | NCT02128269 |
| Acute (GVHD), GIGVHD | Phase 2 | Terminated | NCT02245412 | |
| Healthy | Phase 1 | Completed | NCT01883544 | |
| Healthy | Phase 1 | Completed | NCT01454986 | |
| Zilucoplan, C5-targeting peptide | GMG | Phase 3 | Recruiting | NCT04115293 |
| ALS | Phase 2/3 | Enrolling by invitation | NCT04436497 | |
| ALS | Phase 2/3 | Recruiting | NCT04297683 | |
| GMG | Phase 3 | Recruiting | NCT04225871 | |
| COVID-19 | Phase 2 | Active, not recruiting | NCT04382755 | |
| COVID-19 | Phase 3 | Recruiting | NCT04590586 | |
| IMNM | Phase 2 | Recruiting | NCT04025632 | |
| PNH | Phase 2 | Active, not recruiting | NCT03225287 | |
| PNH | Phase 2 | Completed | NCT03030183 | |
| PNH | Phase 2 | Completed | NCT03078582 | |
| GMG | Phase 2 | Completed | NCT03315130 | |
| Tesidolumab, C5-targeting protein | Wet AMD, Exudative MD | Phase 2 | Completed | NCT01535950 |
| PNH | Phase 2 | Active, not recruiting | NCT02534909 | |
| Transplant associate microangiopathy | Phase 2 | Terminated | NCT02763644 | |
| Kidney transplantation | Phase 1 | Completed | NCT02878616 | |
| Geographic atrophy, AMD | Phase 2 | Completed | NCT01527500 | |
| Advanced AMD | Phase 1 | Completed | NCT01255462 | |
| Geographic atrophy | Phase 1 | Completed | NCT02515942 | |
| Neovascular AMD | Phase 2 | Terminated | NCT01624636 | |
| Non-infectious intermediate uveitis, non-infectious posterior uveitis, non-infectious panuveitis | Phase 2 | Completed | NCT01526889 | |
| Avacopan, C5aR1-targeting small molecule | ANCA-associated vasculitis | Phase 3 | Completed | NCT02994927 |
| ANCA-associated vasculitis | Phase 2 | Completed | NCT02222155 | |
| Vasculitis | Phase 2 | Completed | NCT01363388 | |
| aHUS | Phase 2 | Terminated | NCT02464891 | |
| IgAN | Phase 2 | Completed | NCT02384317 | |
| Hidradenitis, suppurativa acne inversa | Phase 2 | Active, not recruiting | NCT03852472 | |
| C3 Glomerulopathy | Phase 3 | Recruiting | NCT03301467 | |
| Avacincaptad, C5-targeting oligonucleotide | Geographic atrophy secondary to AMD | Phase 2/3 | Completed | NCT02686658 |
| IPCV | Phase 2 | Completed | NCT02397954 | |
| AMD | Phase 2 | Completed | NCT03362190 | |
| IPCV | Phase 2 | Withdrawn | NCT03374670 | |
| Stargardt disease 1 | Phase 2 | Recruiting | NCT03364153 | |
| Geographic atrophysecondary to MD | Phase 3 | Recruiting | NCT04435366 | |
| Avdoralimab, C5aR1-targeting antibody | BP | Phase 2 | Recruiting | NCT04563923 |
| COVID-19, infection advanced and metastatic hematological or solid tumor | Phase 2 | Recruiting | NCT04333914 | |
| COVID-19 | Phase 2 | Active, not recruiting | NCT04371367 | |
| Advanced solid tumors | Phase 1 | Recruiting | NCT03665129 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Giorgio, C.; Zippoli, M.; Cocchiaro, P.; Castelli, V.; Varrassi, G.; Aramini, A.; Allegretti, M.; Brandolini, L.; Cesta, M.C. Emerging Role of C5 Complement Pathway in Peripheral Neuropathies: Current Treatments and Future Perspectives. Biomedicines 2021, 9, 399. https://doi.org/10.3390/biomedicines9040399
Giorgio C, Zippoli M, Cocchiaro P, Castelli V, Varrassi G, Aramini A, Allegretti M, Brandolini L, Cesta MC. Emerging Role of C5 Complement Pathway in Peripheral Neuropathies: Current Treatments and Future Perspectives. Biomedicines. 2021; 9(4):399. https://doi.org/10.3390/biomedicines9040399
Chicago/Turabian StyleGiorgio, Cristina, Mara Zippoli, Pasquale Cocchiaro, Vanessa Castelli, Giustino Varrassi, Andrea Aramini, Marcello Allegretti, Laura Brandolini, and Maria Candida Cesta. 2021. "Emerging Role of C5 Complement Pathway in Peripheral Neuropathies: Current Treatments and Future Perspectives" Biomedicines 9, no. 4: 399. https://doi.org/10.3390/biomedicines9040399