1. Introduction
Spiders of the
Loxosceles genus are commonly referred to as brown spiders and have been broadly implicated in accidents with humans [
1,
2]. In Brazil,
Loxosceles spiders are considered a public health issue, being responsible for approximately 8000 accidents each year over the period of 2017–2019 [
3].
Loxosceles intermedia,
Loxosceles gaucho and
Loxosceles laeta are the most important species from this medical standpoint. Envenomation caused by
Loxosceles spiders generates loxoscelism is the term used to describe the clinical findings observed following the bites, and is categorized into two variants: cutaneous and systemic. Cutaneous loxoscelism is represented by local reactions seen near or at the bite site, such as swelling, erythema, ecchymosis, cutaneous rash, inflammatory response, and dermonecrotic lesion with gravitational spreading. Systemic loxoscelism can result in thrombocytopenia, intravascular hemolysis, acute renal failure, and in some cases, can lead to death in patients [
1,
2,
4].
Loxosceles spiders’ venoms have a colorless appearance and consist of a complex mixture enriched in proteins and glycoproteins with molecular masses ranging from 3–45 kDa [
1,
2,
4]. Amongst the great diversity of toxins found in these venoms, the Phospholipases-D (PLDs) are undoubtedly the most well-studied and functionally well-characterized molecules. PLDs cause the majority of the pathological effects triggered by
Loxosceles crude venoms such as massive inflammatory response and hemolysis, in addition to dermonecrosis, which is the reason why they are also known as dermonecrotic toxins. PLDs present in the venom of
Loxosceles spiders were initially characterized as sphingomyelinases-D based on their ability to cleave sphingomyelin, resulting in choline and ceramide-1-phosphate [
5,
6]. Currently, these molecules are classified as a phospholipases-D due to the wide spectrum of lipids that they can cleave such as lysophosphatidylcholine, lysophosphatidylethanolamine, lysophosphatidylserine, lysophosphatidylinositol, lyso- platelet activating factor (PAF), cyclic phosphatidic acid, in addition to sphingomyelin [
7,
8,
9].
Molecular biology techniques enabled the identification and heterologous expression of several recombinant PLDs isoforms from
Loxosceles spiders’ venoms. These isoforms were characterized and contributed to unveil their participation on the pathophysiology of envenoming.
L. laeta venom gland transcriptome showed that 16.3% of the ESTs encode PLDs [
10]. Two PLDs from
L. laeta, which were named SMase I and SMase II, were recombinantly obtained, and both displayed sphingomyelinase and complement-dependent hemolytic activities, in addition to triggering dermonecrosis in rabbit’s skin [
11,
12]; furthermore, polyclonal antibodies raised against SMase I efficiently neutralized its dermonecrotic activity after toxin and serum were incubated prior to injection in rabbits [
11]. Machado and colleagues [
13] performed proteomic analysis of
L. intermedia,
L. laeta, and
L. gaucho crude venoms by using two-dimensional electrophoresis, revealing spots corresponding to PLDs (30 to 35 kDa); these spots were further analyzed through a combination of mass spectrometry, immunoblotting and chromatography, which resulted in the identification of 11
L. gaucho PLDs isoforms. LgRec1, a
L. gaucho PLD recombinantly obtained, elicited platelet aggregation, induced sphingomyelinase cleavage and prompted direct hemolytic activity; LgRec1 also provoked local reactions near the injection site (i.e., edema and erythema) and dermonecrosis in rabbit’s skin, which were neutralized when LgRec1 was previously incubated with arachnidic antivenom, anti-
L. gaucho, or anti-LgRec1 sera [
14].
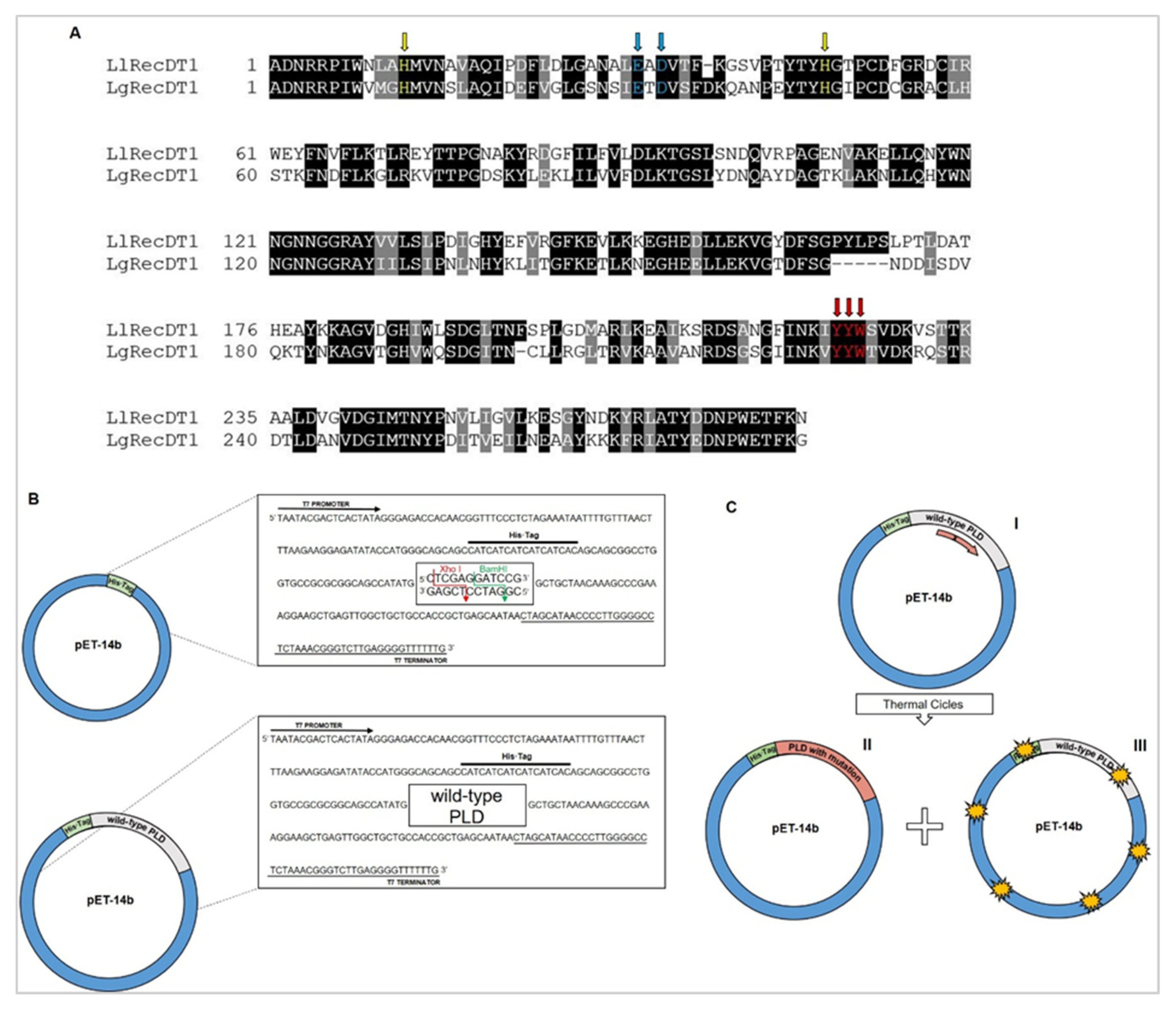
Structural analyses identified PLDs amino acid residues considered important for catalysis, substrate binding, and metal ion binding [
15,
16,
17,
18,
19]. The mechanisms by which these PLDs interact and cleave their substrates have been described and are related to two theoretical models under discussion, which are the cleavage of phospholipids by hydrolysis or by transphosphatidylation [
2,
9,
15]. All of this information is relevant for extending biotechnological applications, such as the use of these molecules in the production of neutralizing antibodies for serum therapy or the diagnosis of loxoscelism [
20,
21,
22].
In this work, we aimed to perform comparative biochemical and biological analyses of wild type and mutants of PLDs from L. laeta and L. gaucho venoms. Results herein obtained may provide rational parameters for the use of recombinant PLDs as antigens for the development of a second-generation anti-Loxosceles serum. In addition, we present relevant data for the production of other therapeutic inputs such as a protective vaccine to be used in areas where envenomation by Loxosceles spiders are epidemic.
2. Materials and Methods
2.1. Wild Type and Site-Directed Mutant PLDs
Wild type LlRecDT1 (GenBank: AY093599.1) and LgRecDT1 (GenBank: JX866729.1) constructions were obtained by using pET-14b plasmids as cloning vectors, according to the methods described by Chaim and colleagues [
23]. Wild type constructions previously mentioned were used for the generation of site-directed mutations H12A-H47A, E32A-D34A, Y228A, and Y228A-Y229A-W230A. Initially, sense oligonucleotides responsible for introducing the intended mutations were designed using the QuikChange Primer Design tool (
https://www.agilent.com/store/primerDesignProgram.jsp (accessed on 7 February 2021)) (
Table 1). The mutated isoforms were produced using pET-14b wild type constructions as templates and the QuikChange
® Multi Site-Directed Mutagenesis Kit (Agilent, Santa Clara, CA, USA) following manufacturers’ instructions. For this, PCR reactions were performed in order to amplify the mutated sequences. Next, amplified sequences were incubated with the endonuclease DpnI, which recognizes and hydrolyzes methylated, non-mutated strands containing the 5′-Gm6ATC-3′ target sequence. All of the mutated constructions were then transformed into chemically competent
Escherichia coli XL10-gold bacterial strain (Agilent, Santa Clara, CA, USA). The colonies were subjected to plasmid mini-preparations using the WizardPlus SV Minipreps DNA Purification Systems
® Kit (Promega, Madison, WI, USA), followed by sequencing reactions using the reagent BigDye
® Terminator v3.1 Cycle Sequencing Kit (Applied Biosystems, Warrington, UK). Each clone was sequenced using the T7 sense primer or the T7 terminator primer by means of automated sequencing in the Applied Biosystems 3500 Genetic Analyzer (Applied Biosystems, Warrington, UK). The sequencing reactions were analyzed using ChromasPro (Version 2.6.4, Technelysium Pty Ltd., Brisbane, QLD, Australia) software to confirm the mutations produced. Comparison between wild type and mutated sequences was carried out using the ClustalW tool (
https://www.genome.jp/tools-bin/clustalw (accessed on 7 February 2021)).
2.2. Recombinant Expression
The constructions containing the site-directed mutations were transformed into
Escherichia coli BL21(DE3)pLysS and plated on Luria–Bertani (LB) agar plates supplemented with ampicillin (100 µg/mL) and chloramphenicol (34 µg/mL). An isolated colony was inoculated into 10 mL of liquid LB broth containing the antibiotics mentioned above at 37 °C for 16 h. Subsequently, the 10-mL culture was inoculated in 1 L of liquid LB containing the antibiotics at 37 °C and the culture growth was monitored until the optical density (OD) at 550 nm reached between 0.4 and 0.6. The isopropyl β-D-thiogalactoside (IPTG) inducer was then added to a final concentration of 0.05 mM and the culture was allowed to grow for additional 3.5 h at 30 °C, under vigorous shaking. Bacterial cells were finally harvested by centrifugation (3500×
g, 10 min) and the resultant pellet was resuspended in 20 mL of binding buffer (50 mM NaH
2PO
4/Na
2HPO
4 pH 8.0, 500 mM NaCl, 10 mM imidazole). After adding lysozyme (1 mg/mL), the suspension was stored at −20 °C [
23,
24].
2.3. Protein Purification
The frozen resuspension was thawed, subjected to mechanical lysis using a French pressure cell press (3 cycles of 5 min on ice-pressure within 5000 and 10,000 psi) and centrifuged (9000×
g, 30 min at 4 °C). The resulting supernatant was collected and 2 mL of Ni
2+-NTA agarose resin was added to it, followed by incubation under gentle agitation for 1 h at 4 °C. The batch was exhaustively washed with washing buffer (50 mM NaH
2PO
4/Na
2HPO
4 pH 8.0, 500 mM NaCl, 20 mM imidazole) until the OD at 280 nm reached 0.01/0 or until the readings remained constant. The recombinant protein was eluted using elution buffer (50 mM NaH
2PO
4/Na
2HPO
4 pH 8.0, 500 mM NaCl, 250 mM imidazole), dialyzed against phosphate-buffered saline (PBS: 100 mM NaCl, 80 mM Na
2HPO
4, 20 mM NaH
2PO
4, pH 7.4) and the purity the eluate was analyzed by 12.5% SDS-PAGE under reducing conditions (β-mercaptoethanol 5%) [
24].
2.4. Circular Dichroism
Circular dichroism spectra were acquired to compare the structure of the recombinant wild type proteins and their mutants. The purified proteins were dialyzed against phosphate buffer (20 mM NaH
2PO
4/Na
2HPO
4 pH 7.4 and 150 mM NaCl) at 4 °C and then diluted in the same buffer to a final concentration of 0.5 mg/mL. The spectra were acquired in a Jasco J-810 spectropolarimeter (Jasco Corporation, Tokyo, Japan) using a 1 mm path-length cuvette. Each spectrum, recorded at 0.5 nm intervals, consists of an average of 8 measurements performed at a rate of 50 nm/min, with a response time of 8 s and a bandwidth of 1 nm. The temperature was maintained constant at 20 °C [
24,
25]. Measurements were performed in triplicate. Secondary structures of the recombinant proteins were predicted using the online tool K2D3 (
http://cbdm-01.zdv.uni-mainz.de/~andrade/k2d3 accessed on 7 February 2021).
2.5. Immunoassays
The interaction between wild type isoforms and mutated toxins with antibodies raised against
L. gaucho or
L. laeta crude venoms produced in rabbits were performed by both western blot and ELISA immunoassays. Initially, protein quantification was performed according to Bradford [
26]. For the western blotting analysis, samples containing 2.5 µg of each protein were submitted to SDS-PAGE under reducing conditions and transferred onto a nitrocellulose membrane using the Trans-Blot
® SD Semi-Dry Electrophoretic Transfer Cell System (Bio-Rad, Hercules, CA, USA) for 30 min at 25 V. The membranes were incubated with hyperimmune sera raised against
L. gaucho or
L. laeta crude venom (1:3000). Control of antivenom sera specificity was performed using pre-immune serum. After incubation with the mentioned sera, membranes were treated with secondary alkaline phosphatase-conjugated anti-IgG antibody (1:5000) (Sigma-Aldrich, St. Louis, MO, USA). Finally, results were developed using BCIP/NBT substrate reaction (Promega, Madison, WI, USA). For the ELISA antibody capture assay, a 96-well microplate (Nunc MaxiSorp, Roskilde, Denmark) was coated with a solution containing 1 μg/mL protein (100 μL per well) in 0.02 M sodium bicarbonate buffer pH 9.6 overnight at 4 °C. After that, the plate was washed with PBS-Tween 20 solution (0.05%), blocked with PBS-casein (2%) for 1 h at 37 °C and the reactions were incubated for 1 h with hyperimmune sera against
L. gaucho or
L. laeta crude venom (1:10,000) at 37 °C. Control of antivenom sera specificity was performed with pre-immune serum. Thereafter, the plate was washed with PBS-Tween 20 solution (0.05%) and wells were incubated with secondary antibodies against IgG-rabbit conjugated with horseradish peroxidase (Sigma Aldrich, San Luis, MO, USA) (1:5000) for 1 h at 37 °C. Finally, the colorimetric reactions were developed using a solution containing 0.4 mg/mL ortho-phenylene diamine (OPD) with 4 μL/mL H
2O
2 in citrate buffer for peroxidase pH 5.0 (50 mM NaH
2PO
4, 24 mM citric acid) for 30 min at 25 °C and reactions were then stopped with 50 μL of 1 M H
2SO
4. Absorbance values were measured at 490 nm in spectrophotometer (Meridian ELX 800) [
23,
27]. Each treatment was assessed in quadruplicate and the results are shown as mean ± SEM of three independent experiments.
2.6. Structural Analyses
The analysis of the three-dimensional structures was performed using the crystallographic structures of the PLDs of
L. intermedia (Protein Data Bank (PDB) ID: 3RLH) and
L. laeta (PDB ID: 2FR9), and the structural models of the mutants built using SWISS-MODEL based on their respective PDB IDs as a template. The native and mutants
L. gaucho PLDs models were constructed using the
L. intermedia PDB ID 3RLH as a template. The electrostatic potential was calculated using the program APBS according to the molecular electrostatic potential ranging from −5.0 to 5.0 [
28] and implemented in PYMOL (version 2.4, GitHub, San Francisco, CA, USA) [
29].
2.7. Sphingomyelinase Activity
Sphingomyelinase activity was determined using the Amplex Red Assay Kit (Thermo Fisher Scientific, Waltham, MA, USA). To measure the sphingomyelinase activity of the recombinant toxins, they were incubated (10 μg) in microtubes containing the Amplex Red reagent mixture in reaction buffer (Tris-HCl 100 mM pH 7.4 containing 10 mM MgCl2), including the substrate sphingomyelin (SM: egg sphingomyelin, chicken). Wild type recombinant isoforms, LlRecDT1 and LgRecDT1, were used as positive controls and reactions containing only the Amplex Red reagent with the substrate SM (without any toxin) were used as negative controls. After incubation at 37 °C for 30 min, the reactions were transferred from the microtubes to a 96-well black fluorimeter plate and the fluorescence was measured on a fluorimeter (Tecan Infinite M200, Männedorf, Switzerland) using excitation wavelength at 540 nm and emission at 570 nm. All treatments were performed in triplicate and the results represent an average of three experiments ± SEM.
2.8. High Performance Thin Layer Chromatography (HPTLC)
The phospholipase activity of the recombinant PLDs was also assessed by HPTLC. For this, samples of 50 μg of each PLD were incubated with 1 mg/mL of sphingomyelin (SM) for 2 h at 37 °C. The organic phase was recovered using 1 mL of a water:butanol solution (1:1), from which 200 μL was collected and dried. Next, the organic phase was resuspended in chloroform and 20 μL was then applied to the base of the thin-layer chromatographic silica plate with capillary tubes. A solution containing chloroform:methanol:methylamine (65:35:10,
v/
v/
v) was used as the mobile phase [
24]. The samples were visualized as blue spots when the plates were sprayed with molybdenum blue dye. The results shown are from one experiment out of two independent experiments that were performed.
Densitometry of the digital images from the HPTLC plates were acquired using the GeneSnap software for G: Box Chemi XL (Syngene, Cambridge, UK) and quantified by the Quantity One software for Chemic Doc XRS (Bio-Rad, Hercules, CA, USA). The percentages of cleavage by LlRecDT1 and LgRecDT1 were considered as 100%. Results represent the densitometry of one experiment.
2.9. Animals
Adult Swiss mice weighing between 25 and 30 g were supplied by the Animal House of the Biological Sciences Center of the Federal University of Paraná and adult New Zealand rabbits weighing approximately 3 kg were acquired from the Canguiri Experimental Farm of Federal University of Parana and kept at Production and Research Center of Immunobiological Products (CPPI, Brazil). Animal experiments were carried out in accordance to the Brazilian Guidelines for Care and Use of Animals for Scientific and Teaching Purposes established by the National Council for Control of Animal Experimentation (CONCEA) and the International Guidelines for animal experimentation. The Ethics Committee for Animal Use from the Biological Sciences Sector of the Federal University of Parana (CEUA/BIO-UFPR) approved all of the protocols using animals in this work (Statement number 1112).
2.10. Hemolytic Activity
The hemolysis assay was conducted as described by Chaves-Moreira and colleagues [
7], with modifications. Initially, 4 mL of rabbit blood was harvested with acidic EDTA Na
+ 5% and centrifuged at 2000×
g for 15 min, followed by aspiration and discard of the overlying platelet-rich plasma and buffy coat. The remaining erythrocytes were washed three times in Tris buffer sucrose (TBS: 250 mM sucrose, 10 mM Tris/HCl, pH 7.4). After washing steps, the erythrocytes were resuspended in TBS with 1 mM CaCl
2 to obtain an initial concentration of 5 × 10
8 cells/mL. Aliquots containing 200 μL of the erythrocyte suspension were placed in microtubes, followed by addition of 200 μL of a solution containing wild type or mutant PLDs (10 μg) solubilized in TBS. Negative control reactions contained only cell suspensions just in TBS and positive control reactions were constituted by cell suspensions in distilled water and Triton X-100 0.1% (
v/
v). The reactions were incubated under gentle and constant agitation at 37 °C. After the treatment times (0, 3, 6, 12, and 24 h), the microtubes were centrifuged at 4 °C using a refrigerated microfuge (Centrifuge 5804 R, Eppendorf, Hamburg, Germany), for 3 min at 3500×
g. The absorbance of free hemoglobin in the supernatant was read at 550 nm in a spectrophotometer (Meridian ELx 800, BioTek Instruments, Winooski, VT, USA). Absorbance readings were converted to percent hemolysis considering the positive control absorbance as 100% lysis. Each treatment was assessed in triplicate and the results are shown as mean ± SEM of three independent experiments performed.
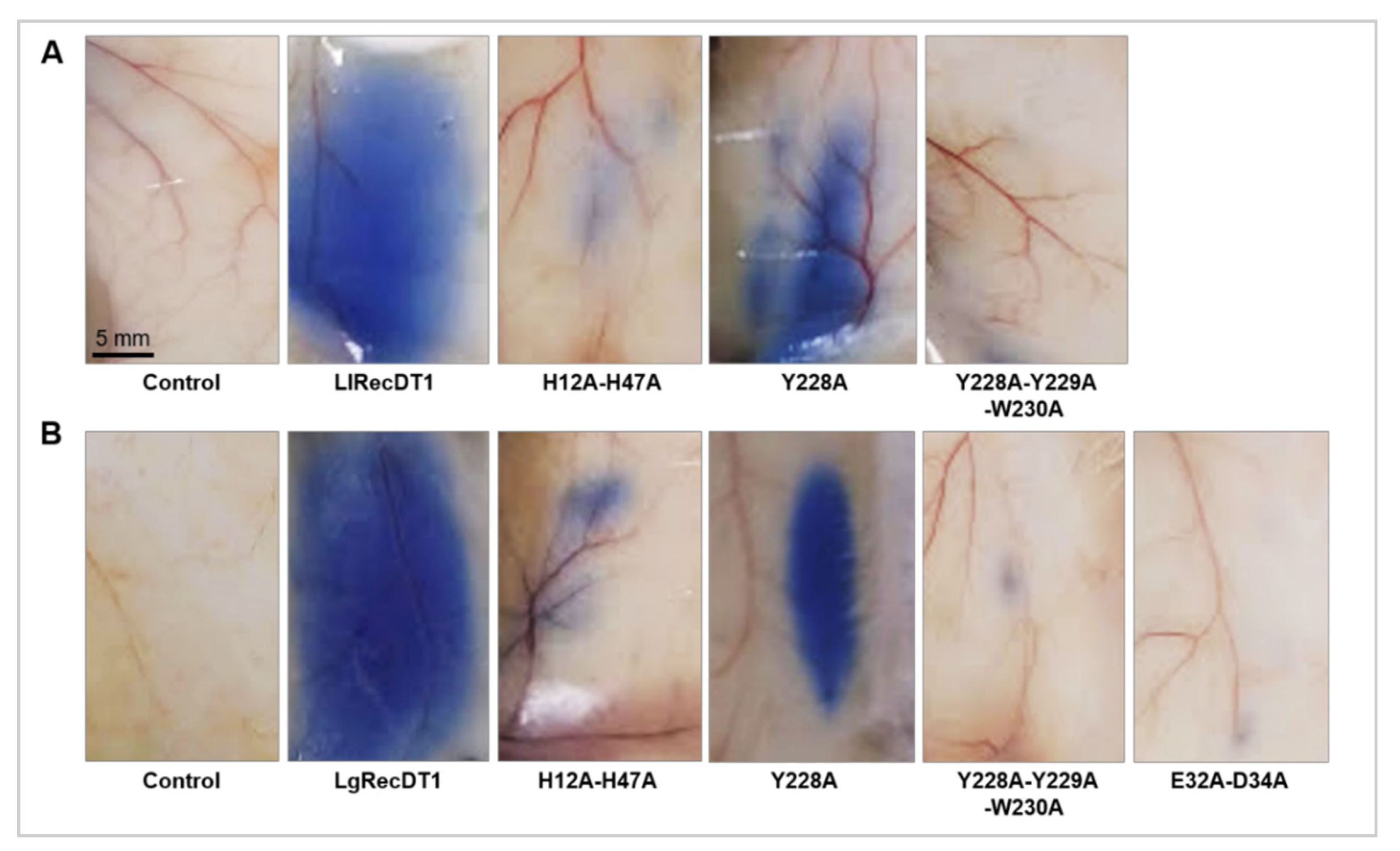
2.11. Vascular Permeability Assay
In order to evaluate alterations triggered by the recombinant PLDs in the microvascular permeability, the Miles assay was performed [
24,
30]. For this analysis, mice were intravenously injected with Evans Blue diluted in PBS (30 mg/kg), followed by intradermal administration of wild type or recombinant mutant PLDs samples (10 μg) 5 min later at the dorsolateral region of the animals. Each treatment group consisted of five animals and the negative control was represented by animals that received the vehicle (PBS) without any toxin. After 1 h, the mice were euthanized using ketamine (30 mg/kg) and xylazine (4 mg/kg) and the dye leakage was visualized (bluish stain). A tissue patch of the dorsal skin from each animal was removed and photographed.
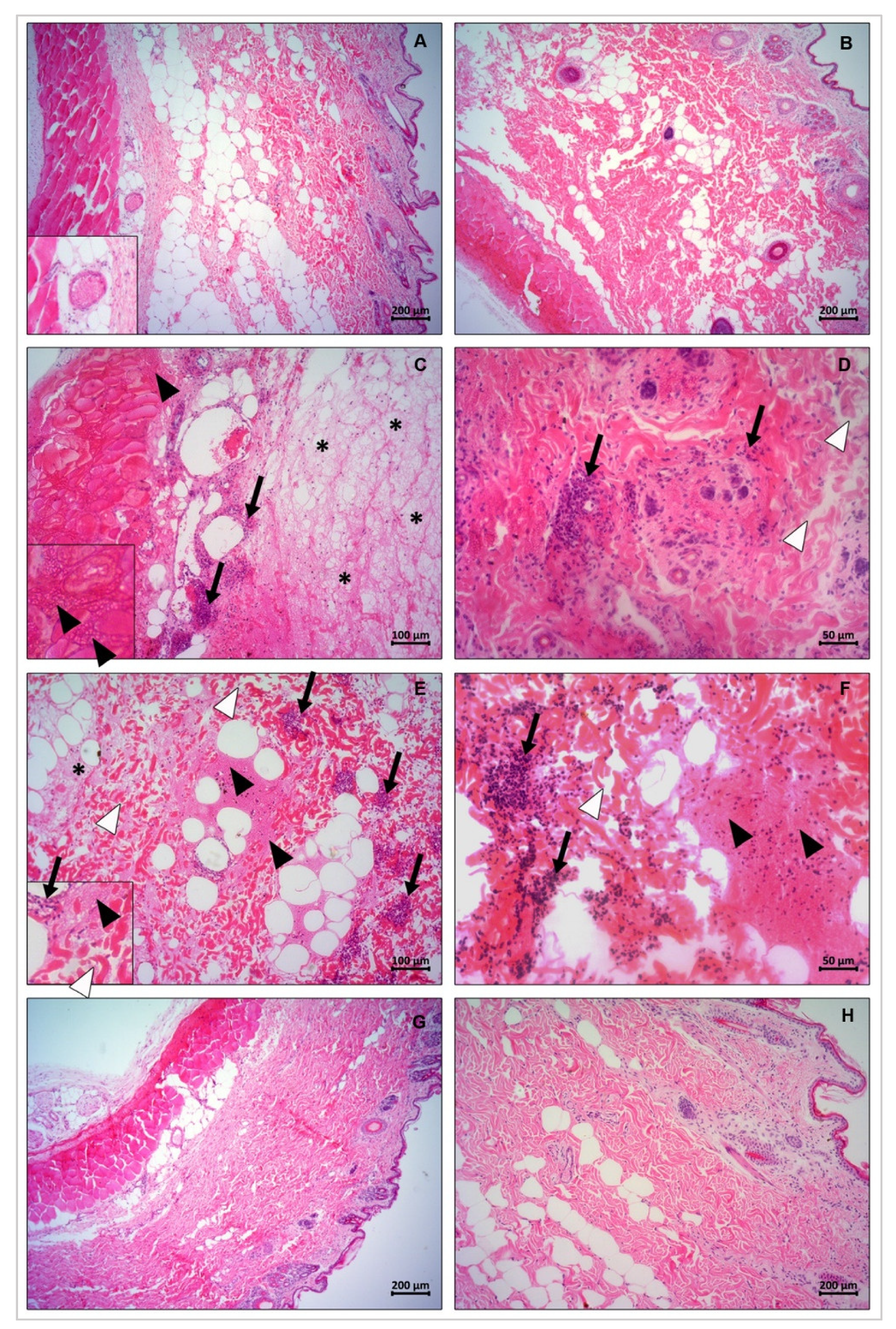
2.12. In Vivo Dermonecrosis
PLDs dermonecrotic activity was evaluated after intradermal application of 10 μg of the wild type and recombinant mutant toxins at a dorsolateral shaven area of rabbits’ skin. PBS solution was administered to animals of the control group. The test was performed in duplicate for each sample. Animals were monitored throughout the evolution of the dermonecrotic reaction and macroscopic images were taken after 0, 3, 6, and 24 h post toxin injection. After the maximum exposure time (24 h), the rabbits were euthanized with ketamine (240 mg/kg) and xylazine (28 mg/kg), followed by tissue excision for histopathological analysis purpose [
24].
2.13. Histological Methods for Light Microscopy
Excised rabbit skin sections were fixed in Alcohol–Formalin–Acetic Acid (ALFAC) fixative solution (ethanol 85%, formaldehyde 10%, and glacial acetic acid 5%) for 16 h at 4 °C, dehydrated in a graded series of ethanol and embedded in paraffin for 2 h at 58 °C. The fixed samples were processed in thin sections (4 μm), placed on glass slides for histological analysis, and stained with hematoxylin and eosin (H&E). The sections were analyzed and photographed under a light microscope Olympus BX41 with DP 72 camera for image capture (Olympus, Tokyo, Japan).
2.14. Statistical Analyses
Values were expressed as mean and standard error of the mean. Statistical analysis of the ELISA and hemolytic assays were performed using two-way analysis of variance (ANOVA) followed by post-hoc Tukey test for average comparisons using the GraphPad Prism 6 program (GraphPad Software, San Diego, CA, USA), considering p value < 0.001 as statistically significant. Statistical analysis of the Sphingomyelinase activity assay was performed using one-way analysis of variance (ANOVA) followed by post-hoc Tukey test for average comparisons using the GraphPad Prism 6 program, considering p value < 0.05 as statistically significant.
4. Discussion
Accidents caused by brown spider bites constitute a public health problem in many parts of the world, especially in South American countries such as Brazil, Argentina, Mexico, Peru, and Chile. In these countries,
Loxosceles spiders are endemic and accidents with them have resulted in critical problems to exposed populations. In the United States of America, especially in the South and Center-South States, many accidents are also reported. In Brazil, these accidents prevail mainly in populations living in the southern states [
1,
2,
34,
35,
36,
37]. The treatment currently established for loxoscelism is essentially symptomatic for the cutaneous form and it aims to halt the robust inflammatory response, which is a key process associated to the pathophysiology of the accidents, as well as the dermonecrosis (aseptic coagulative necrosis). Treatments include steroidal anti-inflammatory drugs such as prednisolone or dapsone, in addition to analgesics as acetylsalicylic acid or paracetamol. Antibiotics, such as erythromycin or cephalosporins, can also be used to treat secondary infections, and antihistamines have also been prescribed to reduce allergic reactions or skin rash [
1,
2,
34,
35,
38,
39,
40]. In the case of systemic loxoscelism, also known as viscero-cutaneous form, treatments include steroidal anti-inflammatory drugs and antivenom serum therapy. In the most severe cases, antivenom serum therapy is widely adopted in countries such as Brazil, Argentina, Mexico, and Peru [
1,
2,
34,
35,
38,
39,
40]. In some regions in Brazil, especially in the southeastern states, the antivenom serum therapy used involves the intravenous application of the polyvalent anti-arachnidic serum produced by the Butantan Institute in São Paulo. This anti-arachnidic serum is obtained by horse immunization with a mixture of the crude venoms from the spiders
Loxosceles gaucho and
Phoneutria nigriventer, as well as the crude venoms from the scorpions
Tityus serrulatus and
Tityus bahiensis. In Brazil’s southern states, the serum therapy is based on the administration of the anti-
Loxosceles serum, which is produced by the Production and Research Center of Immunobiological Products (CPPI) in Paraná state. The anti-
Loxosceles serum is also obtained by horse immunization with the crude venoms from the brown spiders
L. intermedia,
L. laeta, and
L. gaucho, which are the most important species from the medical perspective in South America [
1,
2,
34,
35].
The use of antivenom sera in the treatment of loxoscelism is controversial and it is not a practice performed in the United States of America, for example. The use of anti-
Loxosceles serum therapy in cases of the systemic loxoscelism has been successful, which is supported by experimental and clinical data. A study has already shown that the pediatric mortality decreased after serum therapy [
1,
41]. Besides that, it has also been shown that anti-
Loxosceles serum experimentally administered in rabbits reduced the noxious alterations often seen followed venom exposure, such as hematological disorders, extravascular deposition of fibrinogen and alterations in renal function [
1,
42]. On the other hand, experimental and clinical evidence supports that antivenom serum therapy is ineffective in the treatment of cutaneous loxoscelism for reasons that still remain unknown [
42]. It is speculated that the mentioned ineffectiveness involves the mechanisms following cell/toxin interactions, or simply because patients are slow to seek medical help. The early events of unregulated inflammatory response and dermonecrosis start to develop in the first 4–8 h after the accidents so that a delay in initiating the use of serum therapy no longer results in the neutralization of the venom toxins [
1,
2,
42,
43]. The ineffectiveness of the anti-
Loxosceles serum therapy in cutaneous loxoscelism could be understood based on the cell biology mechanisms associated to the envenoming. One of the possible explanations lies in the inability of the anti-toxins antibodies to neutralize the toxins after activation or induction of molecular changes in the main target-cells (endothelial cells of the blood vessels, fibroblasts, platelets, and erythrocytes) [
20]. Another possibility is that the dermonecrotic toxins (PLDs), which cause almost all of the effects triggered by crude venoms, represent between 15 and 20% of toxin-encoding transcripts in the venom-producing glands. This could generate a low proportional concentration of PLD-neutralizing antibodies in the serum when compared to the high proportional concentration of neutralizing-antibodies for other toxins, resulting in an ineffective serum. If the mentioned hypothesis is true, the production of polyclonal sera with crude venoms enriched with recombinant PLDs or sera obtained using only PLDs as antigens could have a higher neutralizing antibody titer and greater efficiency in serum therapy.
In this work, our main objective was to generate molecular constructions for recombinant expression of PLDs found in the venoms of
L. laeta and
L. gaucho, two species of brown spiders with medical importance and endemic to many South American countries. The mutant isoforms lacking biological activity can be used as antigens in high concentrations to obtain an improved serum or vaccine or in other biotechnological applications. Furthermore, these mutant toxins are valuable biotools that can be used to understand PLDs structure-function relationship. The standardization of the cloning, heterologous recombinant expression and purification processes of the recombinant PLDs described herein is a major step since it shows that these toxins can be easily obtained in large scale. PLDs present in the venoms of
L. laeta,
L. intermedia, and
L. arizonica had their structures unveiled at tridimensional level by crystallography approaches [
9,
15,
31,
32]. These structural data enabled a better understanding of the mechanisms involved in the cleavage of lipid substrates as well as an identification of the amino acid residues located in functionally important intramolecular sites/regions. Based on the data about the amino acid residues involved in PLDs functionality, we designed constructions encoding wild type and mutant PLDs for both
L. laeta and
L. gaucho. The selected mutations comprise amino acid residues located in the catalytic site (H12A-H47A), in the regulatory exosite involved in substrate binding (Y228A and Y228A-Y229A-W230A) and in the magnesium ion coordinating site (E32A-D34A) [
15,
20,
24,
31]. With the exception of E32A-D34A mutation for
L. laeta, all of the designed forms (wild type and mutants) were expressed and purified as soluble proteins (
Figure 2). The production of LlRecDT1 E32A-D34A as a soluble protein was unsuccessful despite the numerous attempts (data not shown). The electrophoretic pattern of all wild type and expressed mutant PLDs showed no changes in the migratory profile (such as drag down, aggregations, or fragmentations) nor significant variations in the expected molecular masses (
Figure 2), which suggests that mutations did not alter the physicochemical behavior of these molecules. In addition, circular dichroism data pointed out no significant changes in the overall structure of the recombinant PLDs (
Figure 3). These data are in line with previous studies that generated mutated isoforms of
Loxosceles intermedia PLDs [
24,
44] sharing the same secondary structures profile or spatial conformation/folding of the wild type forms. The comparative molecular modeling and the analysis of the surface electrostatic charges indicated that there are no significant differences between wild type and mutated isoforms for both PLDs of
L. laeta and
L. gaucho, except for a slight reduction in the surface negative charges of
L. gaucho E32A-E34A (
Figure 4C). These results strengthen the experimental data acquired in circular dichroism technique, pointing out that the spatial molecular structures did not undergo significant changes after mutations.
In the immunoassays, the highest recognition pattern of the recombinant toxins by the sera raised against the native toxins of their counterpart venom, although may result from different affinities of the respective antibodies, suggests the existence of different epitopes between toxins from
L. gaucho and
L. laeta (
Figure 5B–D). These data support the idea that protocols for the production of broad-spectrum neutralizing antivenom sera or protective vaccines using recombinant PLDs as antigens must contain recombinant toxins from both venoms, which can assure high neutralization or protection effectiveness following envenomation, especially in regions where both spider species are endemic such as South America.
One of the major challenges for the production of neutralizing antivenom sera and protective vaccines is the choice of antigens. Considering the production of neutralizing hyperimmune sera in animals, it is important that these antigens do not cause great toxic effects in the inoculated animals. The same mindset should be taken into account for antigens used for vaccines. For this reason, the constructions used in this work were designed to generate antigens that meet all of the desirable requirements, i.e., immunogenicity and low toxicity. As observed by spectrofluorimetry and HTPLC, the mutations H12A-H47A, E32A-D34A, and Y228A-Y229A-W230 were extremely efficient in inactivating the PLDs, resulting in only residual sphingomyelin cleavage. On the other hand, the mutation of a single tyrosine at position 228, for both
L. laeta and
L. gaucho PLDs, was not sufficient to inhibit their biochemical properties, indicating that enzymes containing this mutation are not useful for serum-development. Some previous data in the literature have shown that the biochemical activities of PLDs in spider venoms, such as sphingomyelin or lysophosphatidylcholine cleavage, and biological activities, such as dermonecrosis and hemolysis, are deeply dependent on the catalytic activities of these enzymes. Inactive or artificial mutant isoforms that do not degrade sphingomyelin also do not induce dermonecrosis or hemolysis [
23,
24,
33,
44]. When PLDs act upon sphingomyelin or lysophosphatidylcholine present in the membranes of target cells the lipid mediators formed after enzymatic cleavage constitute biologically active molecules and activate the molecular machinery responsible for the signs and symptoms observed after envenomation [
20,
45,
46]. In this sense, after identifying the inhibition of sphingomyelin-cleavage activity in the mutant isoforms H12A-H47A, E32A-D34A, and Y228A-Y229A-W230, we performed biological analyses regarding well-described activities of these enzymes. The mutated PLDs H12A-H47A, E32A-D34A, and Y228A-Y229A-W230 resulted in loss of hemolytic activity, unchanged capillary permeability, strongly reduced signs of dermonecrosis, and abrogated inflammatory response when compared to the wild type isoforms, strongly supporting a correlation between catalytic activity, dependence on ion metal coordination and substrate binding ability with the noxious effects induced by these enzymes/toxins. These data strongly suggest that the conformational epitopes of mutated isoforms that can act as antigenic determinants have not undergone significant changes, when compared to antigenic determinants of wild type isoforms, enabling any of these mutated variants to be used as antigens in the production of neutralizing hyperimmune sera for treatment of injured victims or of protective vaccines.


 ,
, 










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}