
A Gene-Based Machine Learning Classifier Associated to the Colorectal Adenoma—Carcinoma Sequence

 , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Data Description
- GSE100179: based on GPL17586 Affymetrix Human Transcriptome Array 2.0 platform (Affymetrix; Thermo Fisher Scientific Inc., Waltham, MA, USA). Biopsy samples were collected from 20 healthy colon biopsies (Control), 20 colorectal adenoma (Adenoma) and 20 colorectal cancer (CRC) tissues during routine colonoscopy [11];
- GSE117606: based on GPL25373 Affymetrix HT HG-U133+ PM Array Plate. FFPE samples derived from patients with adenoma, tumor, and adjacent tissues. This set contained 71 CRC, 62 Adenoma, and 65 Control samples [12];
- GSE4183: based on GPL570 Affymetrix Human Genome U133 Plus 2.0 Array. Total RNA was extracted, amplified and biotinylated from frozen colonic biopsies of 15 patients with CRC, 15 with Adenoma, and 8 healthy Controls [13];
- GSE71187: based on GPL6480 Agilent-014850 Whole Human Genome Microarray 4x44K G4112F (Agilent Technologies, Santa Clara, CA). This set contained 99 CRC, 58 Adenoma, and 12 Control samples [14].
2.2. Study Design
- Data preprocessing;
- Differential Expression Analysis;
- Feature Selection;
- Unsupervised learning: Cluster analysis;
- Supervised learning: Comparison of different classifiers;
- Test of the most performing learning model.
2.3. Data Pre-Processing
2.4. Differential Expression Analysis
2.5. Feature Selection
2.6. Unsupervised Learning: Clustering Analysis
2.7. Supervised Learning: Classification Model
2.8. Cross-Validation and Performance Metrics
2.9. Gene Set Enrichment Analysis
3. Results
3.1. Differential Expression Analysis and Feature Selection
3.2. Clustering Analysis
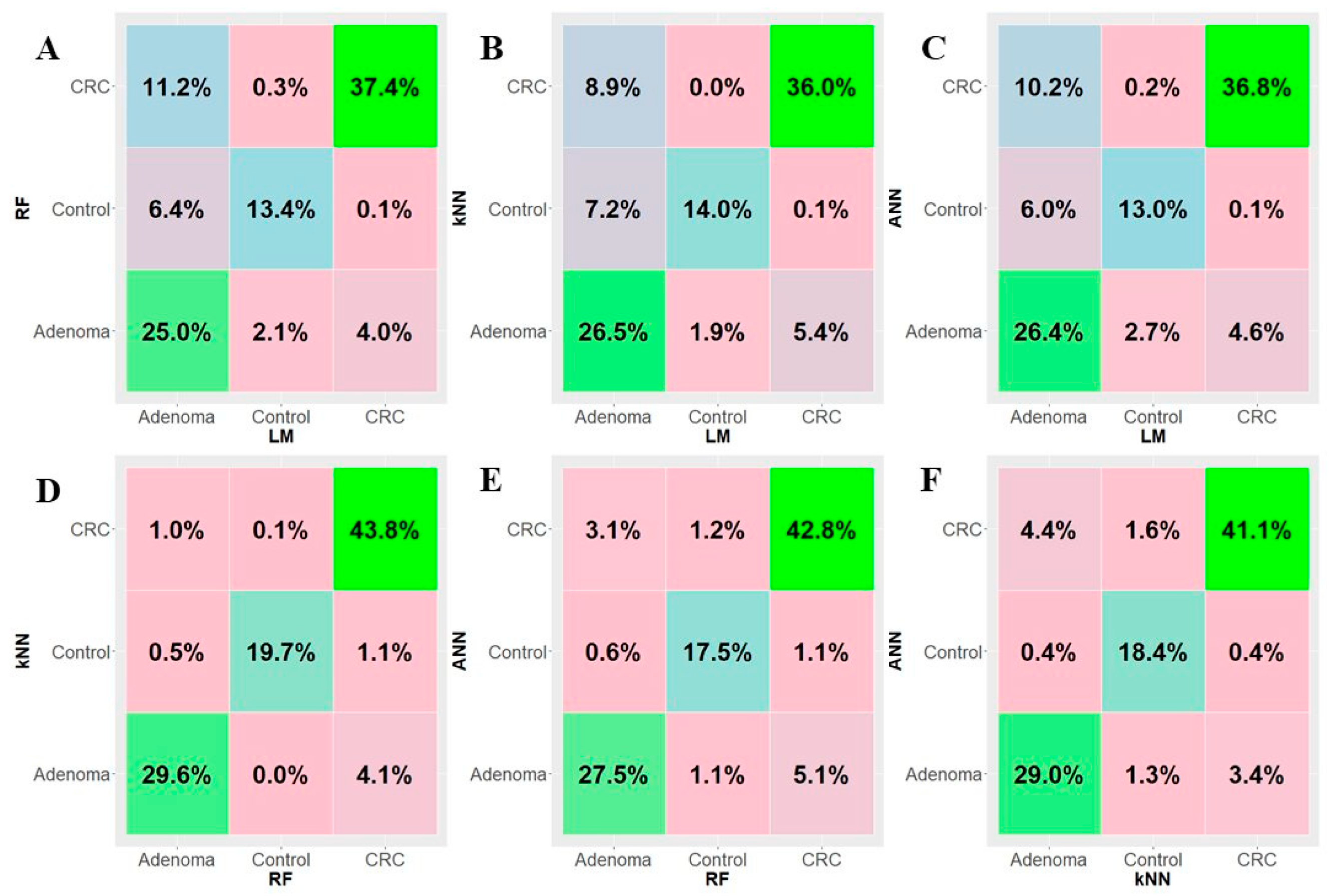
3.3. Machine Learning Analysis and Performance of the Gene-Based Classifier
3.4. Pathway Analysis of the Gene-Based Classifier
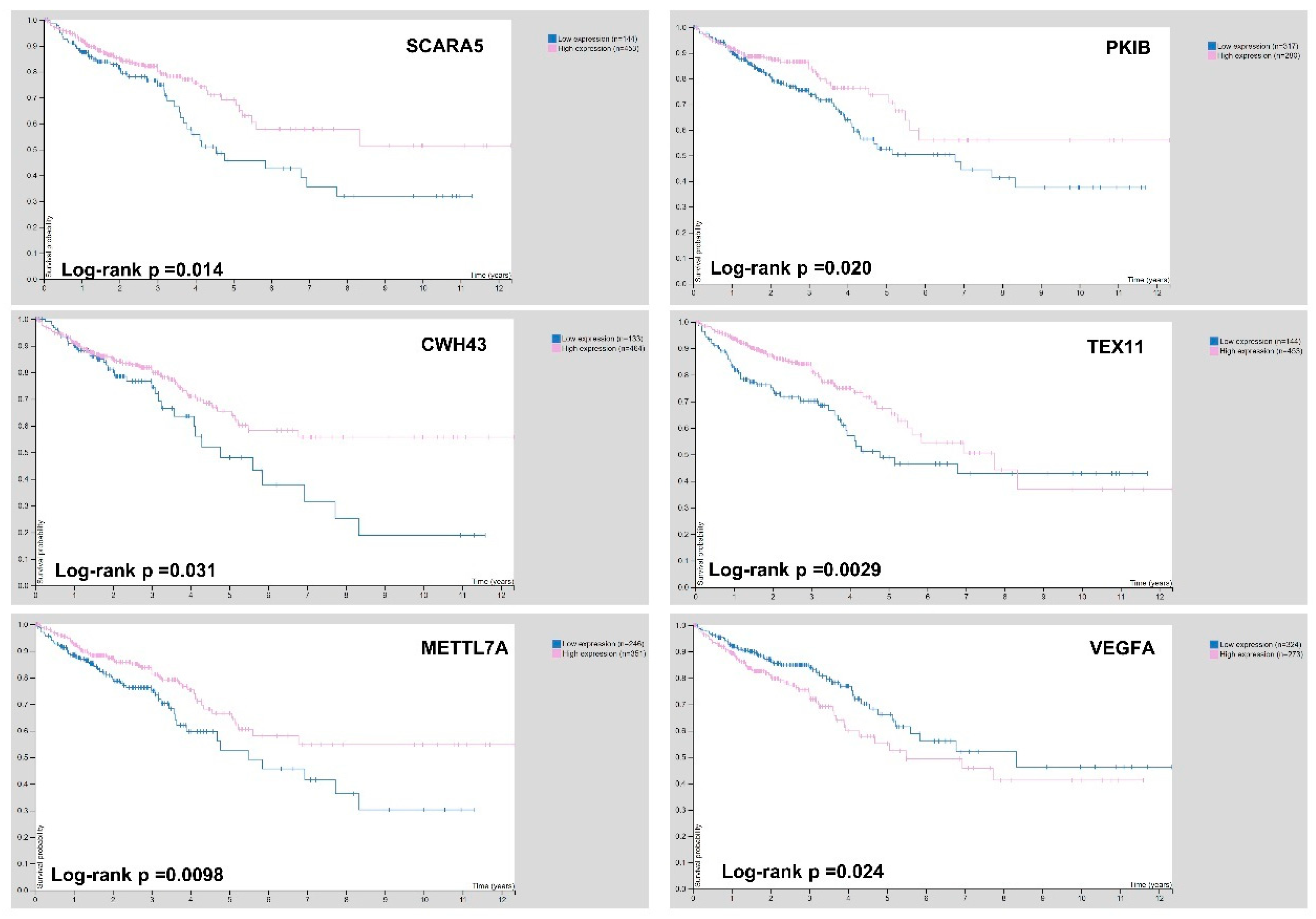
3.5. Selection of Biomarker Genes on Gene Expression and Survival Analysis
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Keum, N.; Giovannucci, E. Global burden of colorectal cancer: Emerging trends, risk factors and prevention strategies. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 713–732. [Google Scholar] [CrossRef] [PubMed]
- Vogelstein, B.; Papadopoulos, N.; Velculescu, V.E.; Zhou, S.; Diaz, L.A., Jr.; Kinzler, K.W. Cancer genome landscapes. Science 2013, 339, 1546–1558. [Google Scholar] [CrossRef]
- Fearon, E.R. Molecular genetics of colorectal cancer. Annu. Rev. Pathol. Mech. Dis. 2011, 6, 479–507. [Google Scholar] [CrossRef] [PubMed]
- Zauber, A.G.; Winawer, S.J.; O’Brien, M.J.; Lansdorp-Vogelaar, I.; van Ballegooijen, M.; Hankey, B.F.; Shi, W.; Bond, J.H.; Schapiro, M.; Panish, J.F.; et al. Colonoscopic polypectomy and long-term prevention of colorectal-cancer deaths. N. Engl. J. Med. 2012, 366, 687–696. [Google Scholar] [CrossRef] [PubMed]
- Corley, D.A.; Levin, T.R.; Doubeni, C.A. Adenoma detection rate and risk of colorectal cancer and death. N. Engl. J. Med. 2014, 370, 1298–1306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaminski, M.F.; Wieszczy, P.; Rupinski, M.; Wojciechowska, U.; Didkowska, J.; Kraszewska, E.; Kobiela, J.; Franczyk, R.; Rupinska, M.; Kocot, B.; et al. Increased rate of adenoma detection associates with reduced risk of colorectal cancer and death. Gastroenterology 2017, 153, 98–105. [Google Scholar] [CrossRef] [Green Version]
- Imperiale, T.F.; Gruber, R.N.; Stump, T.E.; Emmett, T.W.; Monahan, P.O. Performance characteristics of fecal immunochemical tests for colorectal cancer and advanced adenomatous polyps: A systematic review and meta-analysis. Ann. Intern. Med. 2019, 170, 319–329. [Google Scholar] [CrossRef] [Green Version]
- Guinney, J.; Dienstmann, R.; Wang, X.; de Reyniès, A.; Schlicker, A.; Soneson, C.; Marisa, L.; Roepman, P.; Nyamundanda, G.; Angelino, P.; et al. The consensus molecular subtypes of colorectal cancer. Nat. Med. 2015, 21, 1350–1356. [Google Scholar] [CrossRef]
- Kamal, Y.; Schmit, S.L.; Hoehn, H.J.; Amos, C.I.; Frost, H.R. Transcriptomic differences between primary colorectal adenocarcinomas and distant metastases reveal metastatic colorectal cancer subtypes. Cancer Res. 2019, 79, 4227–4241. [Google Scholar] [CrossRef] [Green Version]
- Edgar, R.; Domrachev, M.; Lash, A.E. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002, 30, 207–210. [Google Scholar] [CrossRef] [Green Version]
- Kalmár, A.; Nagy, Z.B.; Galamb, O.; Csabai, I.; Bodor, A.; Wichmann, B.; Valcz, G.; Barták, B.K.; Tulassay, Z.; Igaz, P.; et al. Genome-Wide expression profiling in colorectal cancer focusing on lncRNAs in the adenoma-carcinoma transition. BMC Cancer 2019, 19, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Reumers, J.; Gaj, S.; Van Wesenbeeck, L.; Krishna, V.; Chu, G.; Van den Wyngaert, I.; Verstraeten, K.; Borzillo, G.; Smans, K.; Peeters, P.J.; et al. Gene Expression Data of Patients Presenting with Concurrent Colorectal Adenomas and Colorectal Tumors, Including Adjacent Tissue to the Tumor. 2018. Available online: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE117606 (accessed on 24 May 2021).
- Gyorffy, B.; Molnar, B.; Lage, H.; Szallasi, Z.; Eklund, A.C. Evaluation of microarray preprocessing algorithms based on concordance with RT-PCR in clinical samples. PLoS ONE 2009, 4, e5645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- An, N.; Shi, X.; Zhang, Y.; Lv, N.; Feng, L.; Di, X.; Han, N.; Wang, G.; Cheng, S.; Zhang, K. Discovery of a Novel Immune Gene Signature with Profound Prognostic Value in Colorectal Cancer: A Model of Cooperativity Disorientation Created in the Process from Development to Cancer. PLoS ONE 2015, 10, e0137171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2021. [Google Scholar]
- Carvalho, B.S.; Irizarry, R.A. A Framework for Oligonucleotide Microarray Preprocessing. Bioinformatics 2010, 26, 2363–2367. [Google Scholar] [CrossRef]
- Irizarry, R.A.; Hobbs, B.; Collin, F.; Beazer-Barclay, Y.D.; Antonellis, K.J.; Scherf, U.; Speed, T.P. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics 2003, 4, 249–264. [Google Scholar] [CrossRef] [Green Version]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef] [PubMed]
- Smyth, G.K. Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Stat. Appl Genet. Mol. Biol. 2004, 3, 3. [Google Scholar] [CrossRef]
- Kursa, M.B.; Rudnicki, W.R. Feature selection with the Boruta package. J. Stat. Softw. 2010, 36, 11. [Google Scholar] [CrossRef] [Green Version]
- Ralston, A.; Wilf, H.S. Mathematical Methods for Digital Computers; Technical Report; John Wiley and Sons: New York, NY, USA, 1960. [Google Scholar]
- Breiman, L. Random forests. Mach. Learn. 2001, 45, 5–32. [Google Scholar] [CrossRef] [Green Version]
- Kuhn, M. Caret: Classification and Regression Training; R Package Version 6.0-73. Available online: https://CRAN.R-project.org/package=caret (accessed on 30 June 2021).
- Rousseeuw, P.J. Silhouettes: A graphical aid to the interpretation and validation of cluster analysis. J. Comput. Appl. Math. 1987, 20, 53–65. [Google Scholar] [CrossRef] [Green Version]
- Kriegel, H.-P.; Schubert, E.; Zimek, A. The (black) art of runtime evaluation: Are we comparing algorithms or implementations? Knowl. Inf. Syst. 2017, 52, 341–378. [Google Scholar] [CrossRef]
- Tibshirani, R.; Walther, G.; Hastie, T. Estimating the number of clusters in a data set via the gap statistic. J. R. Stat. Soc. Ser. B 2001, 63, 411–423. [Google Scholar] [CrossRef]
- Ward, J.H., Jr. Hierarchical grouping to optimize an objective function. J. Am. Stat. Assoc. 1963, 58, 236–244. [Google Scholar] [CrossRef]
- Kassambara, A.; Mundt, F. Factoextra: Extract and Visualize the Results of Multivariate Data Analyses (R Package Version 1.0.6). Available online: https://CRAN.R-project.org/package=factoextra (accessed on 30 June 2021).
- Liaw, A.; Wiener, M. Classification and regression by randomForest. R News 2002, 2, 18–22. [Google Scholar]
- Fritsch, S.; Guenther, F.; Guenther, M.F. Package ‘Neuralnet’. In Training of Neural Networks; 2019; Available online: https://github.com/bips-hb/neuralnet (accessed on 30 June 2021).
- Robin, X.; Turck, N.; Hainard, A.; Tiberti, N.; Lisacek, F.; Sanchez, J.-C.; Müller, M. pROC: An open-source package for R and S+ to analyze and compare ROC curves. BMC Bioinform. 2011, 12, 77. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y. MLmetrics: Machine Learning Evaluation Metrics. 2016. Available online: https://cran.rproject.org/package=MLmetrics (accessed on 30 June 2021).
- Kruskal, W.H.; Wallis, W.A. Use of ranks in one-criterion variance analysis. J. Am. Stat. Assoc. 1952, 47, 583–621. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raudvere, U.; Kolberg, L.; Kuzmin, I.; Arak, T.; Adler, P.; Peterson, H.; Vilo, J. g:Profiler: A web server for functional enrichment analysis and conversions of gene lists (2019 update). Nucleic Acids Res. 2019, 47, W191–W198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uhlen, M.; Zhang, C.; Lee, S.; Sjöstedt, E.; Fagerberg, L.; Bidkhori, G.; Benfeitas, R.; Arif, M.; Liu, Z.; Edfors, F.; et al. A pathology atlas of the human cancer transcriptome. Science 2017, 357, eaan2507. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, H.T.; Duong, H.Q. The molecular characteristics of colorectal cancer: Implications for diagnosis and therapy. Oncol. Lett. 2018, 16, 9–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heijink, D.; Kleibeuker, J.H.; Jalving, M.; Boersma-van Ek, W.; Koornstra, J.J.; Wesseling, J.; de Jong, S. Independent induction of caspase-8 and cFLIP expression during colorectal carcinogenesis in sporadic and HNPCC adenomas and carcinomas. Anal. Cell. Pathol. 2007, 29, 409–419. [Google Scholar] [CrossRef] [PubMed]
- Cruz, J.A.; Wishart, D.S. Applications of machine learning in cancer prediction and prognosis. Cancer Inform. 2006, 2, 117693510600200030. [Google Scholar] [CrossRef]
- Niknejad, A.; Petrovic, D. Introduction to computational intelligence techniques and areas of their applications in medicine. Med. Appl. Artif. Intell. 2013, 51, 2113–2119. [Google Scholar]
- Chang, S.-W.; Abdul-Kareem, S.; Merican, A.F.; Zain, R.B. Oral cancer prognosis based on clinicopathologic and genomic markers using a hybrid of feature selection and machine learning methods. BMC Bioinform. 2013, 14, 170. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Zeng, M.L.; Shi, P.C.; Cao, Y.P.; Zhang, J.L.; Xie, Y.P. SCARA5 is a Novel Biomarker in Colorectal Cancer by Comprehensive Analysis. Clin. Lab. 2020, 66. [Google Scholar] [CrossRef]
- You, K.; Su, F.; Liu, L.; Lv, X.; Zhang, J.; Zhang, Y.; Liu, B. SCARA5 plays a critical role in the progression and metastasis of breast cancer by inactivating the ERK1/2, STAT3, and AKT signaling pathways. Mol. Cell. Biochem. 2017, 435, 47–58. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Zheng, D.-L.; Qin, F.-S.; Cheng, N.; Chen, H.; Wan, B.-B.; Wang, Y.-P.; Xiao, H.-S.; Han, Z.-G. Genetic and epigenetic silencing of SCARA5 may contribute to human hepatocellular carcinoma by activating FAK signaling. J. Clin. Investig. 2010, 120, 223–241. [Google Scholar] [CrossRef] [Green Version]
- Dou, P.; Zhang, D.; Cheng, Z.; Zhou, G.; Zhang, L. PKIB promotes cell proliferation and the invasion-metastasis cascade through the PI3K/Akt pathway in NSCLC cells. Exp. Biol. Med. 2016, 241, 1911–1918. [Google Scholar] [CrossRef] [Green Version]
- Laczmanska, I.; Sasiadek, M.; Laczmanski, L. The comparison between molecular tumour profiling in microdissected and surgical tissue samples. Anticancer Res. 2018, 38, 1415–1418. [Google Scholar] [PubMed]
- Wang, P.J.; McCarrey, J.R.; Yang, F.; Page, D.C. An abundance of X-linked genes expressed in spermatogonia. Nat. Genet. 2001, 27, 422–426. [Google Scholar] [CrossRef]
- Yang, F.; Gell, K.; van der Heijden, G.W.; Eckardt, S.; Leu, N.A.; Page, D.C.; Benavente, R.; Her, C.; Höög, C.; McLaughlin, K.J.; et al. Meiotic failure in male mice lacking an X-linked factor. Genes Dev. 2008, 22, 682–691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, T.; Wu, S.; Shen, X.; Li, L. Network cluster analysis of protein–protein interaction network identified biomarker for early onset colorectal cancer. Mol. Biol. Rep. 2013, 40, 6561–6568. [Google Scholar] [CrossRef]
- Feltes, B.C.; Poloni, J.d.F.; Nunes, I.J.G.; Faria, S.S.; Dorn, M. Multi-Approach Bioinformatics Analysis of Curated Omics Data Provides a Gene Expression Panorama for Multiple Cancer Types. Front. Genet. 2020, 11, 586602. [Google Scholar] [CrossRef] [PubMed]
- Jun, F.; Peng, Z.; Zhang, Y.; Shi, D. Quantitative proteomic analysis identifies novel regulators of methotrexate resistance in choriocarcinoma. Gynecol. Oncol. 2020, 157, 268–279. [Google Scholar] [CrossRef]
- Yang, Y.; Zhu, Y.; Li, X.; Zhang, X.; Yu, B. Identification of potential biomarkers and metabolic pathways based on integration of metabolomic and transcriptomic data in the development of breast cancer. Arch. Gynecol. Obstet. 2021, 303, 1599–1606. [Google Scholar] [CrossRef]
- Zhou, S.; Shen, Y.; Zheng, M.; Wang, L.; Che, R.; Hu, W.; Li, P. DNA methylation of METTL7A gene body regulates its transcriptional level in thyroid cancer. Oncotarget 2017, 8, 34652. [Google Scholar] [CrossRef] [Green Version]
- Folkman, J.; D’Amore, P.A. Blood vessel formation: What is its molecular basis? Cell 1996, 87, 1153–1155. [Google Scholar] [CrossRef] [Green Version]
- Carmeliet, P.; Jain, R.K. Angiogenesis in cancer and other diseases. Nature 2000, 407, 249–257. [Google Scholar] [CrossRef]
- Hanahan, D.; Folkman, J. Patterns and emerging mechanisms of the angiogenic switch during tumorigenesis. Cell 1996, 86, 353–364. [Google Scholar] [CrossRef] [Green Version]
- Ferroni, P.; Spila, A.; Martini, F.; D’Alessandro, R.; Mariotti, S.; del Monte, G.; Graziano, P.; Buonomo, O.; Guadagni, F.; Roselli, M. Prognostic value of vascular endothelial growth factor tumor tissue content of colorectal cancer. Oncology 2005, 69, 145–153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grothey, A.; Galanis, E. Targeting angiogenesis: Progress with anti-VEGF treatment with large molecules. Nat. Rev. Clin. Oncol. 2009, 6, 507–518. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cluster Label | ||||
|---|---|---|---|---|
| Adenoma | CRC | Control | ||
| True Label | Adenoma | 139 | 10 | 6 |
| CRC | 36 | 155 | 14 | |
| Control | 1 | 4 | 100 | |
| Accuracy and AUC (%) | ||||
|---|---|---|---|---|
| LM | RF | k-NN | ANN | |
| Accuracy | ||||
| AUC | ||||
| Sensitivity (%) | ||||
| Class | LM | RF | k-NN | ANN |
| Control | ||||
| Adenoma | ||||
| CRC | ||||
| Precision (%) | ||||
| Class | LM | RF | k-NN | ANN |
| Control | ||||
| Adenoma | ||||
| CRC | ||||
| F1 Score (%) | ||||
| Class | LM | RF | k-NN | ANN |
| Control | ||||
| Adenoma | ||||
| CRC | ||||
| Accuracy (%) | ||||
|---|---|---|---|---|
| LM | RF | k-NN | ANN | |
| Accuracy | ||||
| Sensitivity (%) | ||||
| Class | LM | RF | k-NN | ANN |
| Control | ||||
| Adenoma | ||||
| CRC | ||||
| Precision (%) | ||||
| Class | LM | RF | k-NN | ANN |
| Control | ||||
| Adenoma | ||||
| CRC | ||||
| F1 Score (%) | ||||
| Class | LM | RF | k-NN | ANN |
| Control | ||||
| Adenoma | ||||
| CRC | ||||
| Gene Set Name | p-Value | FDR q-Value |
|---|---|---|
| HALLMARK_EPITHELIAL_MESENCHYMAL_TRANSITION | 3.65 × 10−7 | 1.83 × 10−5 |
| HALLMARK_HYPOXIA | 1.76 × 10−4 | 4.41 × 10−3 |
| HALLMARK_ANGIOGENESIS | 1.16 × 10−3 | 1.45 × 10−2 |
| HALLMARK_HEDGEHOG_SIGNALING | 1.16 × 10−3 | 1.45 × 10−2 |
| HALLMARK_IL2_STAT5_SIGNALING | 2.71 × 10−3 | 1.96 × 10−2 |
| HALLMARK_KRAS_SIGNALING_UP | 2.75 × 10−3 | 1.96 × 10−2 |
| HALLMARK_XENOBIOTIC_METABOLISM | 2.75 × 10−3 | 1.96 × 10−2 |
| Term Name | Term ID | T | U | p_adj |
|---|---|---|---|---|
| cell junction | GO:0030054 | 2107 | 18964 | 6.228 × 10−4 |
| microvillus membrane | GO:0031528 | 27 | 18964 | 7.055 × 10−3 |
| cell projection membrane | GO:0031253 | 346 | 18964 | 3.373 × 10−2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lacalamita, A.; Piccinno, E.; Scalavino, V.; Bellotti, R.; Giannelli, G.; Serino, G. A Gene-Based Machine Learning Classifier Associated to the Colorectal Adenoma—Carcinoma Sequence. Biomedicines 2021, 9, 1937. https://doi.org/10.3390/biomedicines9121937
Lacalamita A, Piccinno E, Scalavino V, Bellotti R, Giannelli G, Serino G. A Gene-Based Machine Learning Classifier Associated to the Colorectal Adenoma—Carcinoma Sequence. Biomedicines. 2021; 9(12):1937. https://doi.org/10.3390/biomedicines9121937
Chicago/Turabian StyleLacalamita, Antonio, Emanuele Piccinno, Viviana Scalavino, Roberto Bellotti, Gianluigi Giannelli, and Grazia Serino. 2021. "A Gene-Based Machine Learning Classifier Associated to the Colorectal Adenoma—Carcinoma Sequence" Biomedicines 9, no. 12: 1937. https://doi.org/10.3390/biomedicines9121937